The contrary conservation situations of two local critically endangered species, Vaccinium emarginatum (Ericaceae) and Elatostema platyphyllum (Urticaceae), growing on the eastern edge of the distribution
 Mayu Shibabayashi1*
Mayu Shibabayashi1*  Taiga Shimizu1 Chinatsu Tokuhiro2 Yoshihisa Suyama2
Taiga Shimizu1 Chinatsu Tokuhiro2 Yoshihisa Suyama2  Shota Sakaguchi3 Takuro Ito1
Shota Sakaguchi3 Takuro Ito1  Chih-Chieh Yu4
Chih-Chieh Yu4  Kuo-Fang Chung4,5 Jun’ichi Nagasawa6 Toshiaki Shiuchi7
Kuo-Fang Chung4,5 Jun’ichi Nagasawa6 Toshiaki Shiuchi7  Goro Kokubugata8 Atsushi Abe9 Akiyo Naiki10
Goro Kokubugata8 Atsushi Abe9 Akiyo Naiki10  Atsushi J. Nagano11
Atsushi J. Nagano11  Yuji Isagi1
Yuji Isagi1- 1Graduate School of Agriculture, Kyoto University, Kyoto, Japan
- 2Graduate School of Agricultural Science, Tohoku University, Osaki, Miyagi, Japan
- 3Graduate School of Human and Environmental Studies, Kyoto University, Kyoto, Japan
- 4School of Forestry and Resource Conservation, National Taiwan University, Taipei, Taiwan
- 5Biodiversity Research Center, Academia Sinica, Taipei, Taiwan
- 6Kyoto Botanical Garden, Kyoto, Japan
- 7Botanic Gardens of Toyama, Toyama, Japan
- 8Department of Botany, National Museum of Nature and Science, Tsukuba, Ibaraki, Japan
- 9Okinawa Churashima Research Center, Motobu, Okinawa, Japan
- 10Iriomote Station, Tropical Biosphere Research Center, University of the Ryukyus, Taketomi, Okinawa, Japan
- 11Faculty of Agriculture, Ryukoku University, Kyoto, Japan
As biodiversity loss continues, there is an urgent need to develop efficient conservation measures to protect diversity with limited conservation resources. Conservation targets have generally been selected based on their population size, but more detailed assessments clarifying the phylogenetic genetic status, history, and phylogenetic uniqueness of rare species is crucial to set more appropriate and effective conservation measures. In Japan, the Act on Conservation of Endangered Species of Wild Fauna and Flora designated endangered plants with high conservation priority, but >40% of these species also grow overseas. We conducted comparative analyses based on ddRADseq and MIG-seq to evaluate the population conservation status and value of Vaccinium emarginatum and Elatostema platyphyllum which are growing across national borders at the eastern edge of their species distribution range. The analyses revealed contrasting conservation status between the two species; the Japanese population of V. emarginatum had lower genetic diversity at the individual level and phylogenetically differentiated from Taiwanese populations, while that of E. platyphyllum had higher diversity at the individual level and is a relatively recent migrant with little phylogenetical differentiation from Taiwanese populations. The two species, which share the common feature of being critically rare in Japan, showed contrasting genetic/phylogenetic characteristics. This study provided useful information for appropriate conservation measures based on species’ phylogenetic traits and genetic diversity.
1. Introduction
Despite intensive efforts, biodiversity continues to decline (Rands et al., 2010). The Red List criteria at regional levels developed by the World Conservation Union (IUCN) relates primarily to distribution range, population size, and its fluctuations (Gärdenfors et al., 2001). Although many species at risk are recognized that need to be conserved according to the criteria, conservation resources are limited. Therefore, not only evaluating species at risk based on distribution range or population size, but also clarifying the phylogenetic genetic status, history, and phylogenetic uniqueness of rare species is crucial to its success (Vane-wright et al., 1991).
Decisions on biological conservation are made in administrative areas, while the distribution of species and the ecological and evolutional processes are independent of such areas (Moilanen and Arponen, 2011). Based on the Act on Conservation of Endangered Species of Wild Fauna and Flora, taxa considered endangered due to human impact are designated as “nationally rare species of wild fauna/flora” in Japan. As of January 2022, 198 species of vascular plants were included as targets of this Act (Ministry of the Environment, 2022), and 98 out of the 198 species are non-endemic and grow outside of Japan. Social and political borders may result in significant additional costs to the effective management of natural ecosystems, although local governments and organizations involved in conservation are only responsible for the organisms that grow within their boundaries. The conservation value of rare species that grow within an area under their responsibility depends on the abundance and phylogenetic properties of the same taxa that grow outside the area. If the genetic traits of the targeted populations are unique to the area, conservation efforts should be directed to them. If, however, they do not differ from those outside the area, then conservation resources do not need to be devoted to them as much. Consequently, in such cases, conservation requires information on the genetic and phylogenetic uniqueness of the target population to prioritize in the selection of the target species.
As a case study, we selected two species, Vaccinium emarginatum and Elatostema platyphyllum, which are rare in Japan occurring at the eastern edge of the species distribution, and analyzed their genetic diversity and intraspecific phylogenetic relationships. The results revealed that the two species, both of which have small populations in Japan, were characterized by contrasting genetic/phylogenetic traits.
The first species, Vaccinium emarginatum Hayata (Ericaceae), is an evergreen epiphytic shrub, and the plant height is 20–60 cm. This species is found in Taiwan and Amami Island of Japan. In Japan, this species is sometimes distinguished from the Taiwanese one (Iokawa et al., 2017), called Vaccinium amamianum Hatusima. V. amamianum was first discovered on Amami Island in 1959 and described as a new species in 1962 based on its shorter, dense hairs on the branches, smaller leaves, and terminal recemose inflorescence (Hatusima, 1962). However, Iokawa et al. (2017) considered that the morphological differences between V. emarginatum and V. amamianum were not significant and they should be considered the same species; in addition, molecular phylogenetic analysis indicated that V. amamianum is synonymous with V. emarginatum (Tsutsumi, 2011). In this study, we treat the two taxa identically and refer to V. emarginatum. In 2000, V. emarginatum was listed as a target of the Act on Conservation of Endangered Species of Wild Fauna and Flora of Japan (Ministry of the Environment, 2022), and classified as “Critically Endangered (CR)” in the latest Red List (Ministry of the Environment, 2020). This species grows on the trunks of large trees such as Castanopsis sieboldii (Makino) Hatus., but the number of host trees has decreased due to forest logging, limiting its habitat (Niihara, 2000). The Ministry of Environment of Japan conducted an exhaustive field survey in 2007 and found that only a few individuals grow in a limited area, and the situation was recognized as similar in 2014 (Ministry of the Environment, 2014). On the other hand, it is not threatened with extinction in Taiwan (Taiwan Endemic Species Research Institute, 2021).
The second species, Elatostema platyphyllum Wedd. (Urticaceae), is an evergreen perennial herb that grows on rocks and cliffs along mountain streams. The stem base is woody, and the plant height is 50–100 cm. The species is found in the Himalayas, southern China, Taiwan, and Japan. In Japan, the species grows only on Iriomote Island (Shih et al., 1995), the northeast limit of its distribution range. It is an evergreen perennial plant that reproduces vegetatively through the rhizome. It is also listed as “Endangered (EN)” in the Red List of Japan (Ministry of the Environment, 2020). On the other hand, it is not threatened with extinction in Taiwan (Kokubugata and Kato, 2015).
This study aimed to investigate the genetic/phylogenetic status and determine the conservation value of Japanese populations of these species to propose effective conservation measures. We obtained genome-wide information through multiplexed inter-simple sequence repeat genotyping by sequencing (MIG-seq) (Suyama and Matsuki, 2015) and double digest Restriction site Associated DNA sequencing (ddRADseq) (Peterson et al., 2012) to characterize the phylogenies, genetic structures, and genetic diversity of both species at individual and population level.
2. Materials and methods
2.1. Plant samples and DNA extractions
Six and four V. emarginatum samples from Japan and Taiwan, respectively, were analyzed (Supplementary Table S1; Figures 1A–C). Leaf samples from the Amami population were obtained from ex situ stocks collected from the native habitat on Amami Island and grown in Toyama Botanic Gardens, Kyoto Botanical Garden, and private nurseries on Amami Island. At all these facilities, all plants were originated from separate wild individuals on Amami Island; the plants have not undergone a generational change since collection, and hence retain their genetic information as it was at the time of collection. For E. platyphyllum, leaf samples were collected from plants growing at least 2 m apart from each other, in Iriomote Island and Taiwan (Figures 1A,D,E), respectively. In Iriomote Island, we took 43 samples throughout the distribution area; in Taiwan, 18 and 22 samples were collected from two growing areas (Wulai and Taiping Shan), respectively (Supplementary Table S2). The leaf samples of both species were preserved in silica gel until DNA extraction. Total genomic DNA was extracted from dried leaf material using a CTAB method (Murray and Thompson, 1980). DNA concentration was measured using the Qubit® dsDNA BR Assay Kit (Invitrogen, MA, United States) and adjusted to 15 ng/μL with TE buffer.
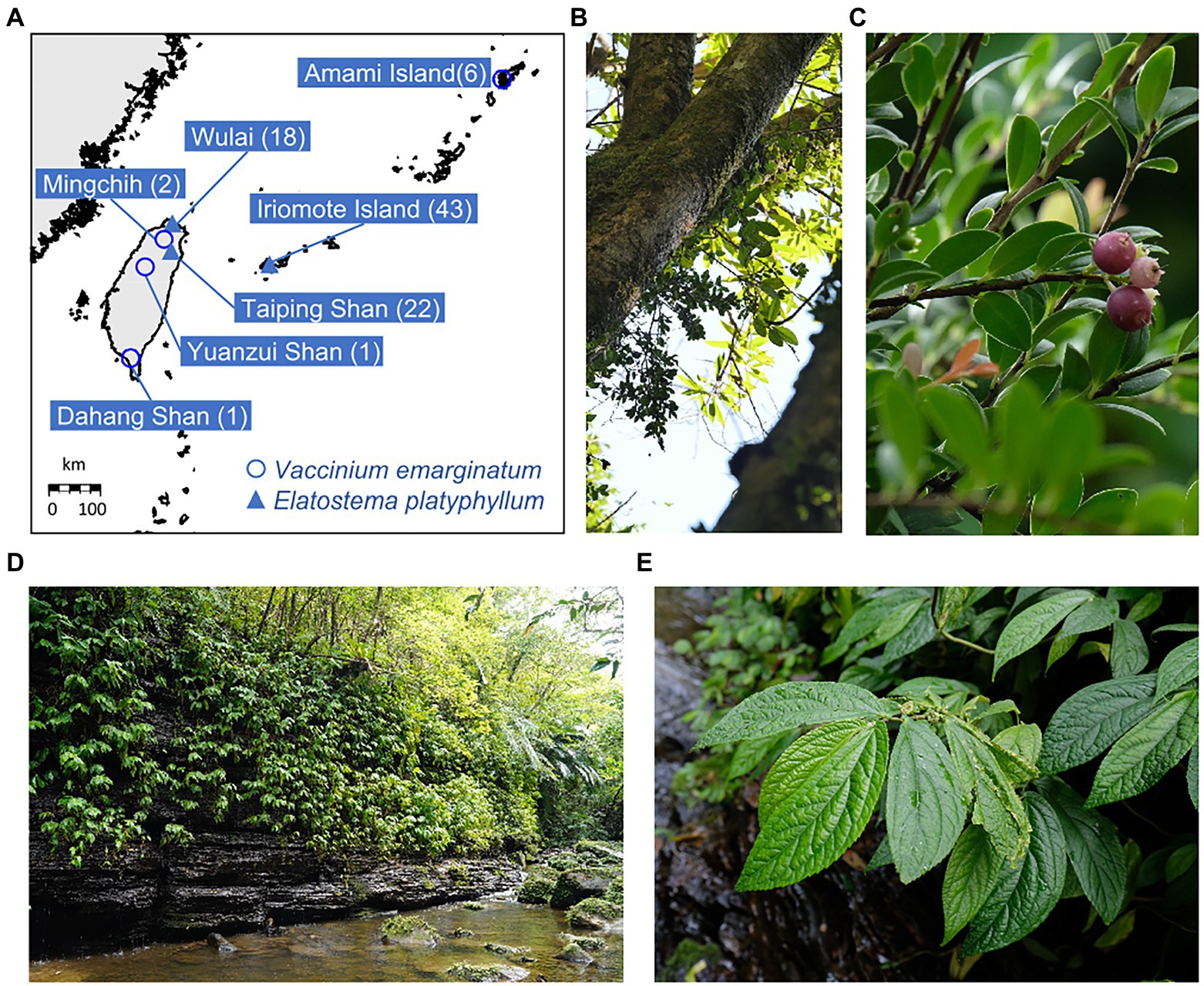
Figure 1. (A) Sampling sites of Vaccinium emarginatum (○) and Elatostema platyphyllum (▲). The numbers of samples are indicated in the parentheses. (B) A habitat of V. emarginatum in Amami Island. This species prefers to grow on the upper part of the trunks. (C) A plant of V. emarginatum producing fruits. (D) The sampling location of E. platyphyllum on Iriomote Island. (E) A plant of E. platyphyllum in its habitat on Iriomote Island.
2.2. MIG-seq, data quality control, and clone identification
Elatostema platyphyllum reproduces vegetatively through the rhizome, therefore, visual identification of individuals is difficult. To identify the clonal samples of E. platyphyllum, all samples of this species were examined using MIG-seq, based on the original protocol with minor modifications (Suyama et al., 2022). A sequencing library of 350–800-bp sequences was diluted to 12 pM and sequenced on an Illumina MiSeq Sequencer (Illumina) using reagents from the MiSeq Reagent Kit v3 (150 cycles, Illumina). Sequencing reads were preprocessed using Trimmomatic version 0.32 (Bolger et al., 2014) to cut two bases off the start and end of reads which are below a threshold quality and align all read lengths to 76 bp. Then, reads with the quality below 30 were removed. Subsequent SNP detection was conducted using the Stacks software package version 2.4 (Rochette et al., 2019). We set the minimum number of identical reads required to create a stack (−m 3), the mismatches allowed between stacks within individuals (−M 2), and maximum distance allowed to align the secondary reads to the primary stacks (–N 4).
The clonal samples were identified using GenoDive version 3.05 (Meirmans, 2020). To distinguish sequence errors and variations between clones, loci data with low missing values retained by 80–100% of the whole samples were used. Additionally, the likelihood of misidentification was reduced by obtaining two independent data from the same samples and removing sequences that did not match between repetitions. Clone assignment was conducted following the manual of GenoDive version 3.05. The Infinite Allele Model was used for the calculation of genetic distance between samples, and missing data were removed from the data set. The threshold to be considered as the same genet was set to 2. The unit identified as the same clone based on the analysis was considered one genet, and 28 samples were used as representative samples for subsequent analysis.
2.3. ddRADseq and data quality control
For phylogenetic and genetic structural analysis, all samples of V. emarginatum and representative samples of E. platyphyllum were used for ddRADseq. Genomic DNA was double digested using the restriction enzymes BglII and EcoRI; the digested DNA fragments and two adapters (BglII adapter and EcoRI adapter) were ligated. Digestion and ligation were performed simultaneously at 37°C for 16 h. The reaction mixture contained 20 ng of genomic DNA, 5 units of BglII (New England BioLabs, Ipswich, MA, United States), 5 units of EcoRI-HF (NEB), 1× NEB buffer2 (NEB), 1× bovine serum albumin (New England BioLabs), 0.2 μM BglII adapter, 0.2 μM EcoRI adapter, 1 mm ATP (Takara, Shiga, Japan), and 300 units of T4 DNA ligase (Enzymatics, Beverly, MA, United States). The reaction product was then purified with AMPure®XP (Beckman Coulter, CA, United States). Purified DNA was amplified by PCR with indexed primers (Sakaguchi et al., 2015) using KOD-Plus-Neo (TOYOBO, Osaka, Japan). PCR product fragments of approximately 320 bp were selected using E-Gel SizeSelect 2% (Life technologies, Waltham, MA, United States). V. emarginatum library was uniquely barcoded and sequenced with 100 bp pair-end reads in two lanes of an Illumina HiSeq2500 (Illumina, San Diego, CA, United States), and that of E. platyphyllum was sequenced with 150 bp pair-end reads in two lanes of an Illumina HiSeqX (Illumina, San Diego, CA, United States) by Macrogen (Seoul, South Korea). Sequencing reads were preprocessed using Trimmomatic version 0.39 (Bolger et al., 2014) to remove adapter sequences and bases with the following parameters: ILLUMINACLIP TruSeq3-PE-2.fa2:30:10 and SLIDINGWINDOW:4:15. Furthermore, all read lengths were aligned to 100 bp for V. emarginatum and 150 bp for E. platyphyllum. The numbers of filtered reads are shown in Supplementary Tables S1, S2.
2.4. SNP calling
The locus assembly and single nucleotide polymorphism (SNP) call was performed using denovo_map.pl. from Stacks version 2.53 (Rochette et al., 2019). We set the minimum number of identical reads required to create a stack (−m 3), the mismatches allowed between stacks within individuals (−M 3), and those between stacks among individuals (−n 2). For E. platyphyllum, the phylogenetic analysis of all samples revealed some identical clones as described below. Since having the same clones in a sample set can introduce biases during SNP calls with stacks, SNP calls were performed again using only one representative sample from the identical clone in the sample set.
2.5. Phylogeny
For phylogenetic analysis, the populations program of the Stacks package was used to extract SNPs and create a variant call format (VCF) file. We set the minimum percentage of individuals in a population required to process a locus (−r 0.8), minimum minor allele frequency (−-min-maf 0.05), and maximum observed heterozygosity required to process a nucleotide site at a locus (−-max-obs-het 0.5). The –write-single-snp option was used to restrict data analysis to only the first SNP per locus to avoid any influence of linkage among SNPs on the following analysis. The resulting VCF files of the SNP extraction were converted to PHYLIP format using PGDSpider version 2.1.1.5 (Lischer and Excoffier, 2012), and the invariant sites were removed with the script ascbias.py.1 Subsequently, a concatenation-based maximum likelihood tree was obtained for each species by RAxML-NG version 0.9.0 (Kozlov et al., 2019), using the best model of evolution determined by MODELTEST-NG (Darriba et al., 2020). Branch support values were obtained through 1,000 standard bootstrap replicates. The resulting phylogenetic tree was drawn with FigTree version 1.4.4 (Rambaut, 2012).
Additionally, the phylogenetic networks were constructed with SplitsTree version 4.10 (Huson and Bryant, 2006). The output VCF files from the populations program of the Stacks package were converted to NEXUS format using the vcf2phylip.py script.2 Subsequently, the networks were constructed using the NEXUS files with default settings in SplitsTree.
2.6. Genetic structure
For genetic structure analysis, the populations program of the Stacks package was used to extract SNPs and create a VCF file. We set the minimum percentage of individuals in a population required to process a locus (−r 0.8), the minimum number of populations a locus must be present (−p 4 for V. emarginatum and 3 for E. platyphyllum), minimum minor allele frequency (−-min-maf 0.05), and maximum observed heterozygosity required to process a nucleotide site at a locus (−-max-obs-het 0.5). Moreover, we used the –write-single-snp option to restrict data analysis only to the first SNP per locus to avoid any influence of linkage among SNPs on the following analysis. This program also calculated the number of private alleles in each population. The VCF files were converted to structure files using PGDSpider version 2.1.1.5.
Genetic structure analysis of the population was performed using the Bayesian clustering method STRUCTURE version 2.3.4 (Pritchard et al., 2000) with K = 1 to 9, 20 times for each K, with 100,000 burn-ins for 100,000 iterations per run. The STRUCTURE Harvester (Earl and VonHoldt, 2012) was used to infer the optimal K based on the delta K method (Evanno et al., 2005). The result of the run with the highest likelihood LnP(D) among all runs for that K was adopted.
In addition, the principal component analysis (PCA) among accessions was carried out using TASSEL version 5 (Bradbury et al., 2007).
2.7. Genetic diversity at the individual and population level
To evaluate genetic diversity at the individual level, we calculated SNP frequency within a single plant, by extracting and outputting SNPs in VCF format using the populations program of the Stacks package with the same options for genetic structure analysis. Then, the Genotype Summary in TASSEL version 5 was used to calculate the percentage of heterozygous SNPs in each individual.
Additionally, ADZE version 1.0 (Szpiech et al., 2008) was used to estimate private allelic richness with a standardized sample size of two, which is the smallest sample size from a single population. In ADZE, the generalized number of private alleles expected in a population is estimated using the rarefaction approach when sample sizes differ across populations (Kalinowski, 2005).
3. Results
3.1. Vaccinium emarginatum
A total of 37,772,894 paired-end reads were obtained from 10 samples, averaging 3,777,289 reads per sample (Supplementary Table S1). We extracted 5,887 SNPs for phylogenetic analysis with RAxML-NG from this dataset. Based on the model select of MODELTEST-NG, the maximum likelihood phylogenetic tree was created using TVM + ASC_LEWIS model. The phylogenetic tree showed that the Japanese and Taiwanese populations formed distinct clades (Figure 2). The Japanese population showed short branches between individuals, indicating small genetic differences within the Japanese population. The phylogenetic network constructed by SplitsTree based on 12,878 SNPs also suggesting the same phylogenetic structure (Supplementary Figure S1).

Figure 2. Phylogenetic analysis based on 5,887 SNPs of V. emarginatum samples using RAxML-NG and visualized by FigTree version 1.4.4. The numbers at the nodes indicate bootstrap support. Sample numbers are color-coded by population; green is for Amami Island, blue is for Mingchih, red is for Dahang Shan and black is for Yuanzui Shan.
For genetic structure analysis, we extracted 4,341 SNPs. The STRUCTURE Harvester analysis showed that delta K peaked at K = 2 (Supplementary Figures S2, S3). Both clustering (Figure 3) and the phylogenetic tree showed a clear genetic differentiation between the Japanese and Taiwanese populations. The result of PCA (Supplementary Figure S4) indicated the differentiation within Taiwanese populations in addition to the genetic structure of Japanese and Taiwanese populations.
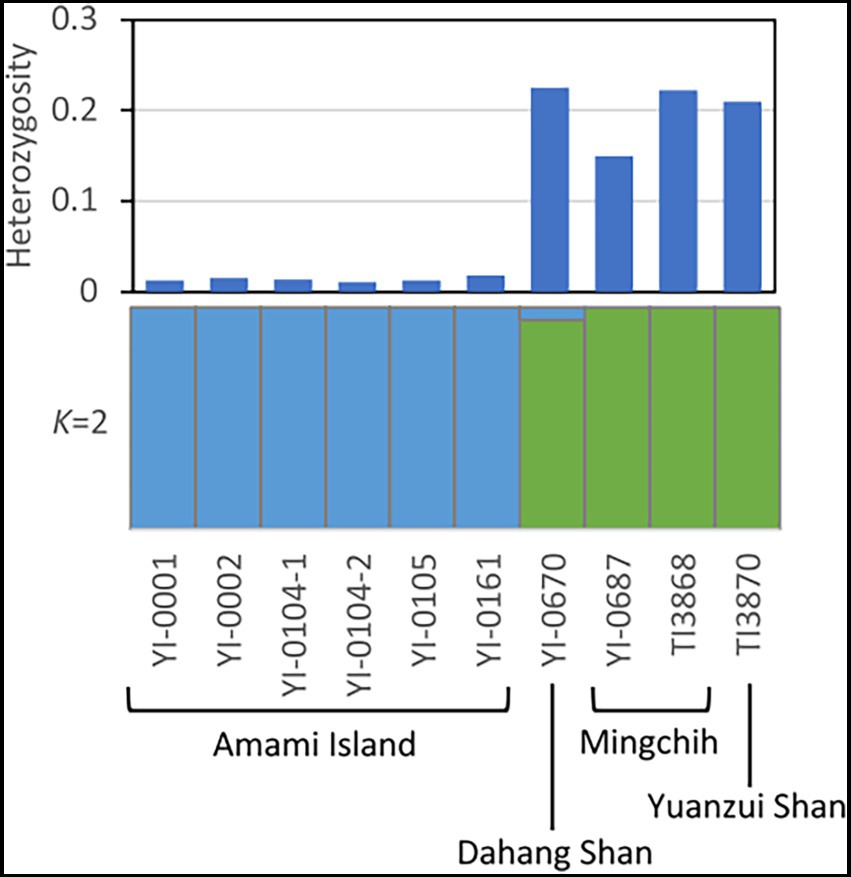
Figure 3. Heterozygosity at individual level and genetic structure analysis (K = 2) of V. emarginatum on genotypes at the 4,341 SNPs. Heterozygosity at individual level was calculated by TASSEL version 5 and genetic clustering was performed by STRUCTURE version 2.3.4.
The individual heterozygosity of the Taiwanese population was 0.202 ± 0.036 on average, while that of the Japanese population was much lower, 0.014 ± 0.003 (t-test, p = 0.000) (Figure 3; Supplementary Table S3). Although the Japanese population had lower heterozygosity, numbers of private allele and private allele richness were larger than Taiwanese samples. The numbers of private alleles in the 4,341 SNPs extracted from Amami Island, Dahang Shan, Mingchih, and Yuanzui Shan samples were 1,188, 727, 569, and 220, respectively. The private allelic richness of each was 0.245 ± 0.006, 0.184 ± 0.005, 0.089 ± 0.004, and 0.083 ± 0.004, respectively.
3.2. Elatostema platyphyllum
An average of 397,838 reads per samples was obtained by MIG-seq and clonal identification was conducted using 144 SNPs. The results showed that the Iriomote population was considered to be composed of one genet and the samples from Taiwan consisted of 27 genets (Supplementary Table S2). One sample from each genet was selected as representative and examined using ddRADseq for the following analysis.
A total of 100,705,390 paired-end reads, averaging 3,596,621 reads per sample, were obtained from the representative 28 samples by ddRADseq (Supplementary Table S2). From the dataset, 3,539 SNPs were extracted for phylogenetic analysis with RAxML-NG. Based on the model select of MODELTEST-NG, the maximum likelihood phylogenetic tree was created using SYM + G4 + ASC_LEWIS model. Within the resulting phylogenetic tree (Figure 4), the individual from Iriomote and Taiping Shan were in the same clade, while the Wulai population and others were clearly separated. The divergence between clades was smaller to that within populations, and the differentiation between the Japan and Taiwan populations was not clear. The phylogenetic network constructed by SplitsTree based on 6,867 SNPs also showed the same situation (Supplementary Figure S5).
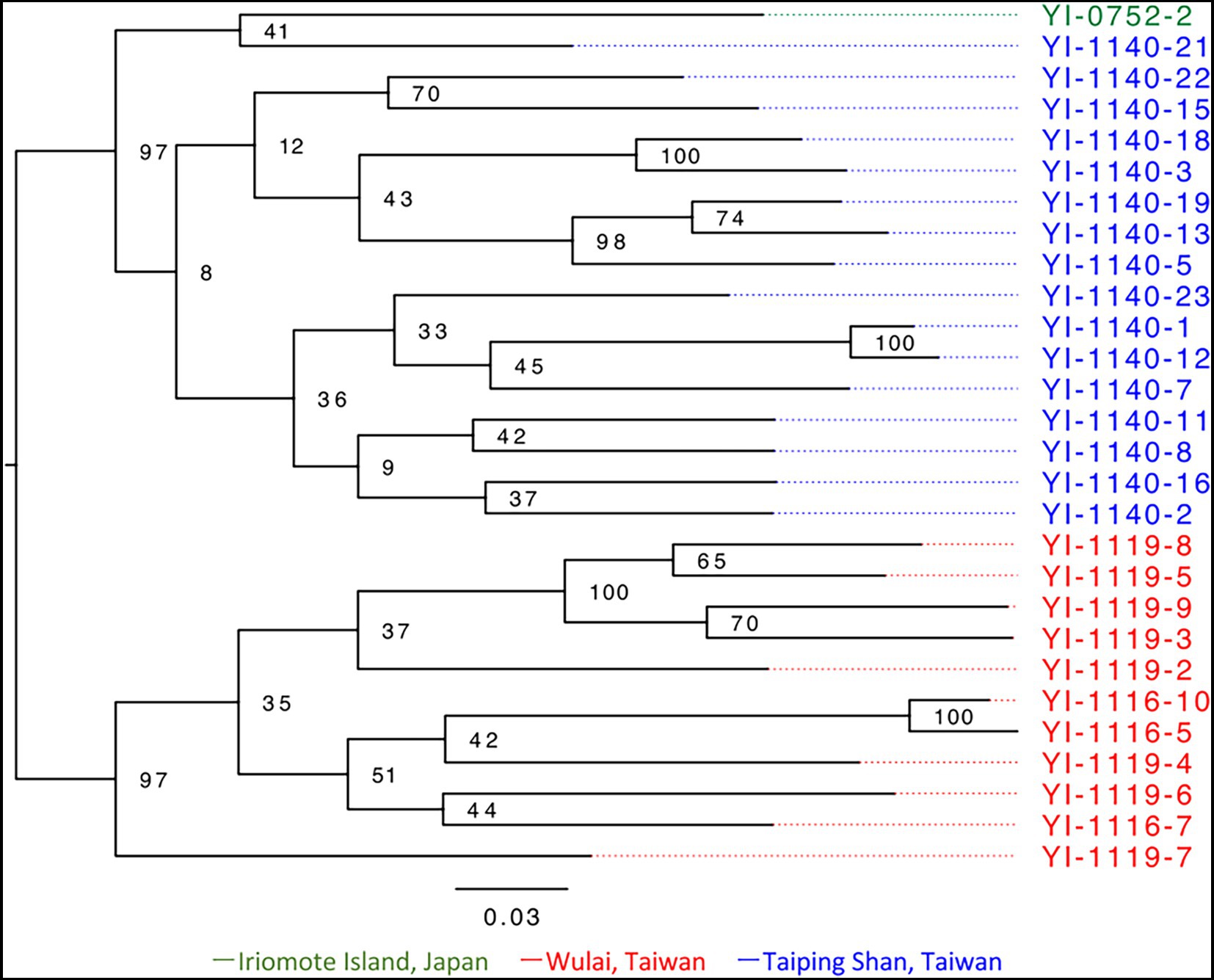
Figure 4. Phylogenetic analysis based on 3,539 SNPs of E. platyphyllum samples which represents the genet groups. This figure was generated by RAxML-NG and visualized by FigTree version 1.4.4. The numbers at the nodes indicate bootstrap support. Sample numbers are color-coded by population; green is for Iriomote Island, red is for Wulai, and blue is for Taiping Shan.
The estimated optimal K value calculated on the extracted 3,404 SNPs was two (Supplementary Figures S6, S7). This result indicated the differentiation between the two populations in Taiwan, but no clear genetic structure over Japanese and Taiwanese populations (Figure 5). This feature is consistent with the architecture in the phylogenetic tree and the result of PCA (Supplementary Figure S8), where differences between samples were relatively larger than differences between clades.
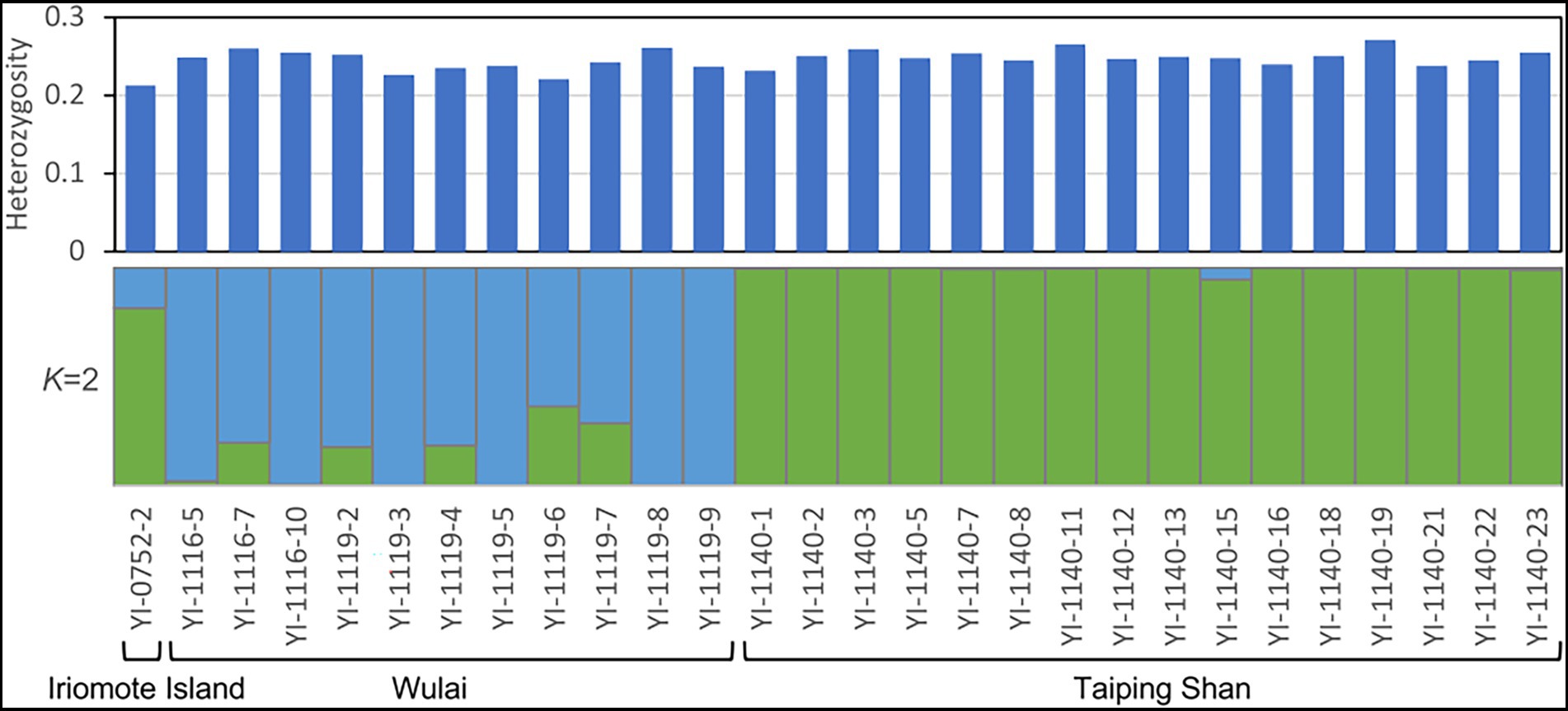
Figure 5. Heterozygosity at individual level and genetic structure analysis (K = 2) of E. platyphyllum on genotypes at the 3,404 SNPs. Heterozygosity at individual level was calculated by TASSEL version 5 and genetic clustering was performed by STRUCTURE version 2.3.4.
The individual heterozygosity of the Japanese sample was 0.214 (standard deviation is not shown because all samples were an identical clone and the representative sample was only one for the Japanese samples), and that of the Taiwanese samples averaged 0.247 ± 0.012 (Figure 5; Supplementary Table S3). The values for the Japanese sample were only slightly lower than those for the Taiwanese sample. Of the extracted 3,404 SNPs, the private alleles were not found in Iriomote Island, while 241 and 349 were found in Wulai and Taiping Shan populations, respectively. The private allelic richness of each population was 0.115 ± 0.004 in Iriomote, 0.159 ± 0.003 in Wulai and 0.141 ± 0.003 in Taiping Shan.
4. Discussion
4.1. Genetic situation and conservation strategy of Vaccinium emarginatum
Phylogenetic and genetic structure analysis revealed much smaller differentiation within the Japanese population of V. emarginatum than in Taiwan (Figures 2, 3). Furthermore, the frequency of heterozygous sites at the individual level in the Japanese population was extremely lower compared to that of the Taiwanese population (Figure 3). Since small populations with low genetic diversity may reduce the population’s survival abilities (Willi et al., 2006), the Japanese population is in a critical situation because of its low genetic diversity at the population and individual levels.
Although Tsutsumi (2011) revealed similarities between samples from Japan and Taiwan in nuclear Internal transcribed spacer and chloroplast sequences, the present analysis based on ddRADseq indicates clear genetic differentiation between Japanese and Taiwanese populations (Figures 2, 3). Furthermore, of the 4,341 loci extracted, we found that the Amami population had 1,188 private alleles. The private allelic richness was also the highest in the Amami population. This indicates that the differences between the Amami and Taiwanese populations in phylogenetic analysis were not only due to lower heterozygosity of Amami population, but also due to the own genetic variations in the Amami population, suggesting the genetic uniqueness. From a conservation point of view, genetically different intraspecific populations require separate genetic management (Moritz, 1995). Therefore, the Japanese and Taiwanese populations should be managed as separate conservation units.
The heterozygosity of populations and individuals decreases more rapidly over generations in small populations (Frankham et al., 2002); the much smaller heterozygosity in Japanese individuals suggests that the Amami population remained small during repeated generational changes. The species was discovered in 1959 in Amami Island, and the low individual heterozygosity of the sample collected in 1959 (YI-0001) indicates that genetic diversity was already low at the time of discovery. It is unlikely that the species was introduced to Amami Island by humans, since it grows on the tops of the trunks of tall trees growing in primary forests that have not been affected by human activities. The habitat of V. emarginatum on Amami Island is highly pristine, with few naturalized plants. Because of its long generation time, it is considered that the Japanese individuals had lost genetic diversity over a long time before the discovery in 1959. The lower heterozygosity of the Amami population can lead to expression of recessive mutations that reduce fitness (Keller and Waller, 2002; Charlesworth and Willis, 2009), which can have a negative impact on the population’s viability.
Although the Amami Island has been registered a UNESCO World Heritage site since 2021, and its nature is being protected more than ever before. Continuous monitoring of the species’ wild habitat is necessary to prevent further reduction of the number of trees on which the species grows.
4.2. Genetic situation and conservation strategy of Elatostema platyphyllum
Elatostema platyphyllum has characteristics that contrast with V. emarginatum. First, the population of E. platyphyllum on Iriomote Island consisted of a single clone. Second, there were no significant phylogenetic differences between the Japanese and Taiwanese samples (Figure 4) and the STRUCTURE analysis also showed no clear genetic structure between them (Figure 5). Therefore, the Japanese population of E. platyphyllum had lower phylogenetic uniqueness than that of V. emarginatum. Third, only slight differences in heterozygosity were observed between Japanese and Taiwanese individuals. Although sexual reproduction and generational changes among a small number of individuals reduce genetic diversity, the reduction was not observed in the Japanese population despite the small number of genet (a single clone). Therefore, it is considered that the Japanese population originated from a single or a small number of individuals immigrated from Taiwan to Iriomote Island in the relatively near past, and has been maintained by vegetative reproduction or with few generational changes. Silvertown (2008) suggested high frequency of clonality in populations of rare or endangered species, of alien plants, or at the edges of the species’ geographical ranges. The Japanese population meets all these conditions.
The Iriomote population is located in the central part of the island, where indigenous vegetation has not been influenced by human activity, and the population seems derived from natural distribution. However, its history is likely as short as a few generations, which is almost equivalent to transient naturalized plants.
5. Conclusion
Despite their small numbers and low population genetic diversity in Japan, V. emarginatum and E. platyphyllum exhibited contrasting characteristics in terms of individual genetic diversity, phylogenetic uniqueness, and the number of generations in Japan. Briefly, the Japanese population of V. emarginatum showed phylogenetic uniqueness, a long history with repeated generational changes, and higher conservation value, while the Japanese E. platyphyllum maintained genetic diversity at individual level, had small phylogenetic difference to the Taiwanese population and had undergone smaller number of generational changes after its immigration to Japan.
Although the conservation status of a species is assessed primarily by the size of population and habitat, the genetic analysis in this study provided other important information to consider. The cases of the two species targeted in this study clearly demonstrate that the status of rare species is characterized not only by their abundance and habitat size, but also from a genetic perspective. In Japan, additional species are planned to be targeted for conservation by the Act on Conservation of Endangered Species of Wild Fauna and Flora, and more intensive conservation management is attempting to be implemented for those species. However, the genetic traits of these Japanese species and populations are not yet fully uncovered. Given the constraints on the resources available for biodiversity conservation, in order to conserve a large number of species at risk efficiently and appropriately, it is imperative to understand the status of the species through multifaceted evaluation and analysis based on genetic information.
Data availability statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession numbers can be found below: NCBI; DRA015126, DRA015424, and PRJNA921842.
Author contributions
MS and YI conceived the study and revised the manuscript critically for important intellectual content. SS, TI, C-CY, K-FC, JN, T. Shiuchi, GK, AA, AN, and YI conducted the field sampling. MS, CT, and AJN performed the experiments. MS, T. Shimizu, CT, and YS analyzed the data. MS wrote the manuscript. All authors read and approved the final manuscript.
Funding
This research was supported by the Environment Research and Technology Development Fund (4-1902 and 4MF-2202) from the Environmental Restoration and Conservation Agency, Japan. Some computations were performed on the supercomputer at the Research Organization of Information and Systems, National Institute of Genetics.
Acknowledgments
We thank H. Yamashita, S. Ichikawa, and T. Hashimoto for their assistance in surveying the habitat conditions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1093321/full#supplementary-material
Footnotes
References
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Charlesworth, D., and Willis, J. H. (2009). The genetics of inbreeding depression. Nat. Rev. Genet. 10, 783–796. doi: 10.1038/nrg2664
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294. doi: 10.1093/molbev/msz189
Earl, D. A., and Vonholdt, B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294x.2005.02553.x
Frankham, R., Ballou, S. E. J. D., Briscoe, D. A., and Ballou, J. D. (2002). Introduction to Conservation Genetics, Cambridge: Cambridge University Press.
Gärdenfors, U., Hilton-Taylor, C., Mace, G. M., and Rodriguez, J. P. (2001). The application of IUCN red list criteria at regional levels. Conserv. Biol. 15, 1206–1212. doi: 10.1111/j.1523-1739.2001.00112.x
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Iokawa, Y., Kurashige, Y., and Takahashi, H. (2017). “Ericaceae” in Wild Flowers of Japan. eds. H. Ohashi, Y. Kadota, H. Kihara, J. Murata, and H. Yonekura, vol. 4 (Tokyo: Heibonsha), 224–262.
Kalinowski, S. T. (2005). Hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 5, 187–189. doi: 10.1111/j.1471-8286.2004.00845.x
Keller, L. F., and Waller, D. M. (2002). Inbreeding effects in wild populations. Trends Ecol. Evol. 17, 230–241. doi: 10.1016/s0169-5347(02)02489-8
Kokubugata, G., and Kato, M. (2015). Global red list of Japanese threatened plants. Avilable at: http://www.kahaku.go.jp/english/research/db/botany/redlist/index.html (Accessed October 10, 2022).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Lischer, H. E., and Excoffier, L. (2012). PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299. doi: 10.1093/bioinformatics/btr642
Meirmans, P. G. (2020). GENODIVE version 3.0: easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol. Ecol. Resour. 20, 1126–1131. doi: 10.1111/1755-0998.13145
Ministry of the Environment (2014). Red Data Book 2014: Threatened Wildlife of Japan (in Japanese), vol. 8. Tokyo: GYOSEI Corporation.
Ministry of the Environment. (2020). Ministry of the Environment Red List (in Japanese). Available at: https://www.env.go.jp/press/107905.html (Accessed August 17, 2022).
Ministry of the Environment. (2022). List of domestic rare wild Fauna and Flora (in Japanese). Available at: https://www.env.go.jp/nature/kisho/domestic/list.html (Accessed August 17, 2022).
Moilanen, A., and Arponen, A. (2011). Administrative regions in conservation: balancing local priorities with regional to global preferences in spatial planning. Biological Conservation 144, 1719–1725. doi: 10.1016/j.biocon.2011.03.00
Moritz, C. (1995). Uses of molecular phylogenies for conservation. Philosophical transactions of the Royal Society of London. Series B Biol. Sci. 349, 113–118. doi: 10.1098/rstb.1995.0097
Murray, M., and Thompson, W. (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8, 4321–4326. doi: 10.1093/nar/8.19.4321
Niihara, S. (2000). Living collection of woody plants endemic to Kagoshima prefecture, southern Japan I (in Japanese). Bull. Kagoshima Pref. Exp. Stn. 5, 19–31.
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S., and Hoekstra, H. E. (2012). Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7:e37135. doi: 10.1371/journal.pone.0037135
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
Rambaut, A. (2012). FigTree v1.4.4. Avilable at: http://tree.bio.ed.ac.uk/software/figtree/
Rands, M. R., Adams, W. M., Bennun, L., Butchart, S. H., Clements, A., Coomes, D., et al. (2010). Biodiversity conservation: challenges beyond 2010. Science 329, 1298–1303. doi: 10.1126/science.1189138
Rochette, N. C., Rivera-Colón, A. G., and Catchen, J. M. (2019). Stacks 2: analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 28, 4737–4754. doi: 10.1111/mec.15253
Sakaguchi, S., Sugino, T., Tsumura, Y., Ito, M., Crisp, M. D., Bowman, D. M., et al. (2015). High-throughput linkage mapping of Australian white cypress pine (Callitris glaucophylla) and map transferability to related species. Tree Genet. Genomes 11, 1–12. doi: 10.1007/s11295-015-0944-0
Shih, B., Yang, Y., Liu, H., and Lu, S. (1995). Notes on Urticaceae of Taiwan. Bot. Bull. Acad. Sinica 36, 155–168.
Silvertown, J. (2008). The evolutionary maintenance of sexual reproduction: evidence from the ecological distribution of asexual reproduction in clonal plants. Int. J. Plant Sci. 169, 157–168. doi: 10.1086/523357
Suyama, Y., Hirota, S. K., Matsuo, A., Tsunamoto, Y., Mitsuyuki, C., Shimura, A., et al. (2022). Complementary combination of multiplex high-throughput DNA sequencing for molecular phylogeny. Ecol. Res. 37, 171–181. doi: 10.1111/1440-1703.12270
Suyama, Y., and Matsuki, Y. (2015). MIG-seq: an effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 5:16963. doi: 10.1038/srep16963
Szpiech, Z. A., Jakobsson, M., and Rosenberg, N. A. (2008). ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24, 2498–2504. doi: 10.1093/bioinformatics/btn478
Taiwan Endemic Species Research Institute. (2021). Taiwan biodiversity network TBN home page (in Taiwanese). Available at: https://www.tbn.org.tw/ (Accessed October 10, 2022).
Tsutsumi, C. (2011). The phylogenetic positions of four endangered Vaccinium species in Japan. Ser. B Bot. 37, 79–86.
Vane-Wright, R. I., Humphries, C. J., and Williams, P. H. (1991). What to protect?—systematics and the agony of choice. Biol. Conserv. 55, 235–254. doi: 10.1016/0006-3207(91)90030-D
Keywords: conservation value, endangered species, phylogeny, genetic diversity, genetic differentiation, genetic structure, SNPs
Citation: Shibabayashi M, Shimizu T, Tokuhiro C, Suyama Y, Sakaguchi S, Ito T, Yu C-C, Chung K-F, Nagasawa J, Shiuchi T, Kokubugata G, Abe A, Naiki A, Nagano AJ and Isagi Y (2023) The contrary conservation situations of two local critically endangered species, Vaccinium emarginatum (Ericaceae) and Elatostema platyphyllum (Urticaceae), growing on the eastern edge of the distribution. Front. Ecol. Evol. 11:1093321. doi: 10.3389/fevo.2023.1093321
Edited by:
Adriano Sfriso, Ca' Foscari University of Venice, ItalyReviewed by:
Joanna Freeland, Trent University, CanadaPrakash Pradhan, West Bengal Biodiversity Board, Government of West Bengal, India
Copyright © 2023 Shibabayashi, Shimizu, Tokuhiro, Suyama, Sakaguchi, Ito, Yu, Chung, Nagasawa, Shiuchi, Kokubugata, Abe, Naiki, Nagano and Isagi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mayu Shibabayashi,  shibaba49@gmail.com
shibaba49@gmail.com