Culex quinquefasciatus Holobiont: A Fungal Metagenomic Approach
 Guillermo A. M. Flores
Guillermo A. M. Flores Rocio P. Lopez
Rocio P. Lopez Carolina S. Cerrudo
Carolina S. Cerrudo V. Fabiana Consolo
V. Fabiana Consolo Corina M. Berón
Corina M. Berón- 1Instituto de Investigaciones en Biodiversidad y Biotecnología (INBIOTEC) - Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Fundación para Investigaciones Biológicas Aplicadas (FIBA), Mar del Plata, Buenos Aires, Argentina
- 2Laboratorio de Ingeniería Genética y Biología Celular y Molecular (LIGBCM), Area Virosis de Insectos (AVI), Departamento Ciencia y Tecnología, Universidad Nacional de Quilmes and CONICET, Bernal, Argentina
Microorganisms associated with mosquitoes have fundamental roles, not only in their nutrition, but also in physiological and immunological processes, and in their adaptation to the environment as well. Studies on mosquito hologenomes have increased significantly during the last years, achieving important advances in the characterization of the “core bacteriome” of some species of health importance. However, the fungal mycobiome has not been exhaustively researched, especially throughout the life cycle of some hematophagous mosquito species. In this work, the diversity and composition of fungal communities in different developmental stages, sexes, and adult nutrition of Culex quinquefasciatus reared on laboratory conditions were characterized, using internal transcribed spacer high throughput amplicon sequencing. Larvae presented a higher fungal richness, while sucrose-fed males and females showed a similar diversity between them. Blood-fed females presented few operational taxonomic units with an even distribution. Results are consistent with the reduction of larval microbiota after molting, observed for the bacterial microbiome in other mosquito species. The filamentous Ascomycota Penicillium polonicum and Cladosporium sp. were present in all stages of the mosquitoes; in addition, the presence of yeasts in the insects or their subsequent colonization associated with their diet is also discussed. These results suggest that some species of fungi could be essential for the nutrition and development of mosquitoes throughout their life cycle.
Introduction
Studies on hematophagous mosquitoes that feed on human blood are central in public health since these insects are capable of vectorizing a variety of pathogens, many of them potentially deadly for more than half of the world’s population (WHO, 2020). In particular, Culex quinquefasciatus is widely distributed in the tropical and subtropical areas in almost all the world, having an important role in the transmission of lymphatic filariasis, West Nile virus, Saint Louis encephalitis virus, the Japanese encephalitis virus (JEV), and other pathogens important to wildlife and human health (Vinogradova, 2000; Becker et al., 2020; Harvey-Samuel et al., 2021).
In recent years there has been an increase in research on mosquito-microbiota interaction, highlighting the role of some microorganisms in mechanisms such as the exploitation of nutrients, obtaining metabolites with physiological or immunological functions, resulting in great adaptability to different environments, in defense against stress agents, and in vector competence, as well (Dong et al., 2009; Dennison et al., 2014; Guégan et al., 2018). It has been demonstrated that the same bacterium can affect distinct mosquito populations in different ways. For instance, the endosymbiont bacteria Wolbachia, can modify the pathogen transmission of agents like dengue virus in Aedes aegypti lines artificially carrying it (Hoffmann et al., 2011), or the human malaria parasite, Plasmodium falciparum, in Anopheles stephensi (Bian et al., 2013). However, in natural mosquito populations, the presence of Wolbachia can be beneficial for their efficient development, affecting its susceptibility against some mosquito pathogens (Díaz-Nieto et al., 2021; Gil et al., 2021). It has also been seen that the density of intracellular Wolbachia has been found to be higher in insecticide resistant mosquito populations (Duron et al., 2006). This clearly shows that the interactions can be multiple, depending on the microorganism-host relationship, type and density of the microorganisms, interactions with the environment, insect nutrition and many other factors, in extremely complex systems. Know and characterize the mosquito holobiont, identifying the ecological community of symbiotic, pathogenic and commensal microorganisms, associated with the mosquitoes body and the assembly of genetic information between them, will allow to understand the complex and coordinated co-evolution of these forms of life (Guerrero et al., 2013; Guégan et al., 2018). On the other hand, it permits the manipulation of the microbiota to control mosquitoes or the transmission of the etiological agents that they vector (Ricci et al., 2011; Huang et al., 2020; Singh and Das, 2020).
At least part of the microorganisms that colonize mosquitoes are acquired during their immature stages in their aquatic habitat, where the larval stages filter organic matter in suspension (Zouache et al., 2022). The microbiota of mosquitoes reared in natural habitats is highly variable and can vary according to their geographic origin, ecological niche, species, sex, and food source (Yun et al., 2014; Coon et al., 2016). At the same time, the microbial density can be influenced by the type of diet ingested by the insect, which depends directly on the stage of development in which it is found and its sex. As it has been shown, the microbiota composition and distribution in Ae. aegypti midgut differs from blood digestion (Gusmão et al., 2010); and in Aedes albopictus differences in the active microbiota between males and females are identified after being fed on fructose solution (Guégan et al., 2020b). Despite this variability, it has been determined, at least for bacteria and fungi, that they generally contain a core microbiome dominated by a small number of taxa (Guégan et al., 2018; Luis et al., 2019).
Although progress has been made in understanding the mosquito holobiont, its fungal composition is not known in detail. In a few Aedes and Culex species, Ascomycota and Basidiomycota are detected, including yeasts and filamentous species, with high prevalence of some particular fungus (Malassigné et al., 2020). In this context, some yeasts, including Wickerhamomyces anomalus (Pichia anomala) have been associated with An. stephensi, Anopheles gambiae, Ae. aegypti and Ae. albopictus and proposed as promising species to implement in methods for the control of mosquito-borne diseases (Ricci et al., 2011). Most recently, fungal microbiota communities, associated with some mosquitoes of the Culex and Aedes genera, were identified through next generation sequencing (Luis et al., 2019; Guégan et al., 2020a). However, the understanding about fungal diversity in mosquitoes and its relationship with other symbiotic microorganisms is very scarce. Therefore, the aim of this work was to identify the fungal community in the Cx. quinquefasciatus holobiont from insectary population throughout the mosquito life cycle, comparing the different stages of development, between the sexes in adults, taking into account the nutritional source between males, females fed on sucrose and females fed on vertebrate blood, using high-throughput amplicon sequencing of the fungal internal transcribed spacer (ITS2) fragment.
Materials and Methods
Ethics Statement
Protocols for blood feeding mosquitoes on mice were reviewed and approved by the Animal Experimental Committee at the Faculty of Exact and Natural Sciences, Mar del Plata University (Institutional Committee on Care and Use of Experimental Animals (CICUAL) N° 2555-04-14). Mice were handled in strict accordance with National Health Service and Food Quality (SENASA) guidelines (Argentina) following the 2011 revised form of The Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health.
Mosquito Rearing and Collection
Larvae and adults of Cx. quinquefasciatus were obtained from the insectary of the Biological Control Laboratory of the INBIOTEC-CONICET, FIBA (Argentina). Insects were reared at standard temperature and relative humidity conditions (24°C, 80 ± 5%) and a 12 h light: 12 h dark photoperiod. Larvae were raised in dechlorinated water and fed with commercial fish food (Shulet Carassius), male and female adults were provided with 20% (w/v) sucrose solution and gravid females were allowed to feed blood from mice. Adults were killed by freezing and wings and legs were removed under Nikon SMZ800 stereoscope.
Mosquito DNA Extraction
A total of 80 individuals (20 insects for each group) were used for the total genomic DNA extraction: 1) third instar larvae 2) sucrose fed male adults 3) sucrose-fed female adults and 4) mice-blood-fed female adults. All sucrose solution-fed adults were processed at 48 h post emergence, and blood-fed females were processed 48 h post blood-feeding (96 h post emergence, after blood digestion). Each insect pool (in duplicates) was surface sterilized with 70% ethanol for 1 min, followed by 3 to 4 washes with sterile phosphate-buffered saline solution (PBS), for 1 min each time. Samples were frozen on liquid nitrogen and homogenized with the DNA extraction kit lysis buffer (DNeasy Blood & Tissue kit, QIAGEN), using glass beads and a teflon tip in a mechanical cell lyser equipment. This process was carried out in four cycles of freezing/drilling, 1 min each. The homogenates were incubated with 2 mg/ml of proteinase K during 3 h for protein degradation, then centrifuged at 12000 rpm. After that, the lysate was transferred to the kit column, following the DNA extraction, according to the manufacturer’s protocol.
Metagenome Amplicon Sequencing
Metagenome amplicon sequencing of the fungal ribosomal RNA gene was performed using gDNA isolated from each sample. Extracted DNA was quantified by picogreen method using Victor 3 fluorometry (Invitrogen, Waltham, MA, USA) and quality condition assessed by gel electrophoresis method. From each mosquito group, sequencing libraries for nuclear ribosomal internal transcribed spacer (ITS2) were constructed. ITS3 (5´- GCATCGATGAAGAACGCAGC -3´) and ITS4 (5´- TCCTCCGCTTATTGATATGC -3´) primers (Bellemain et al., 2010) were used on an Illumina Miseq 300 bp PE platform, in accordance with Illumina 18S rRNA metagenomic sequencing library protocols (Macrogen Inc., Korea).
Sequence Analysis and Taxonomic Assignment
The quality of the paired-end reads in .fastq files was initially checked by FastQC. Further, with a combination of UPARSE-pipeline (Edgar, 2013) and mothur software, applying quality and quimera removing filters, with a minimum length overlap of 50, a maximum number of mismatches in the overlap region of 12; the reads longer than 250 bp with an expected error < 1 were clustered into operational taxonomic units (OTUs) at a level of 97% similarity. Taxonomic assignment was performed on representative sequences from each OTU using massBLASTer and SH mappings of the UNITE database (Nilsson et al., 2019). Unclassified OTUs were taxonomically identified by performing Blast searches against the GenBank standard database, Nucleotide collection (nr/nt), (Supplementary Figure S1).
Bioinformatic Analysis
To analyze sequencing depth in each mosquito group, rarefaction curves with vegan package 2.5 (Oksanen et al., 2020) were constructed. Alpha-diversity indexes to describe the communities diversity were calculated and measured in two categories for each group: richness and diversity. Margalef, Fisher_alpha, Shannon, Inverse Simpson index (InvSimpson) and Sorensen’s indexes were computed using mothur (Schloss et al., 2009) and vegan package 2.5, with a randomization of 100 runs and sampling with replacement. Thus, the Shannon index quantifies species diversity for each collected sample, combining measures of richness and evenness. Margalef index is a specific richness index, Fisher_alpha index applied to richness estimation, is theoretically independent of sample size, and InvSimpson represent the inverse of the probability that two randomly selected individuals in the habitat will belong to the same species. Relative abundance was calculated, and identified taxa were analyzed with Krona (Ondov et al., 2011).
Fungal community composition from mosquito instar populations (ß-diversity) were primarily analyzed using non-metric multidimensional scaling (NMDS) plot by R software v.4.1.2. (RC Team, 2021). The distance matrix was computed based on Bray-Curtis and Euclidean distance analysis with vegan package 2.5, these distances were used to construct NMDS and principal component analysis (PCA) respectively. The PCA was performed in the Galaxy web platform usegalaxy.org (Afgan et al., 2018); with a combination of ggplot2 and ggfortify. Plots were made on Graphpad Prism 8.0.1.
Results
Fungal Species Diversity in Cx. quinquefasciatus
Mosquito Fungal Alpha Diversity
In order to explore the fungal diversity throughout the Cx. quinquefasciatus development, amplicons of the ITS2, from the ribosomal DNA region, were sequenced. A total of 1,509,358 paired-end reads were obtained from gDNA of larvae, males and females fed on sucrose, and females fed on mice blood (Supplementary Table S1). After quality filtering, 34,603 reads were assigned to 55 fungal OTUs at 97% identity (Supplementary Table S2 and Supplementary Data Sheet 1). Rarefaction curves showed that the sequencing depth was sufficient to cover the fungal diversity observed in the mosquito groups (Supplementary Figure S2). Only for the blood-fed females sample the plotted curve did not reach a plateau, indicating that the fungal community was not well represented.
Diversity estimators, Shannon and InvSimpson indices (Table 1 and Figure 1) which measure the richness and homogeneity of the samples, considering the evenness of the species relative abundance, showed similar values for all mosquito groups. Neither of the two indices allow to obtain a comparable diversity value between the mosquito stages. This is because the stages with the highest number of OTUs present heterogeneity and dominance of one species, while the sample with the least number of OTUs (blood-fed females) presents homogeneity (it has few species, but each one has similar relative abundance).
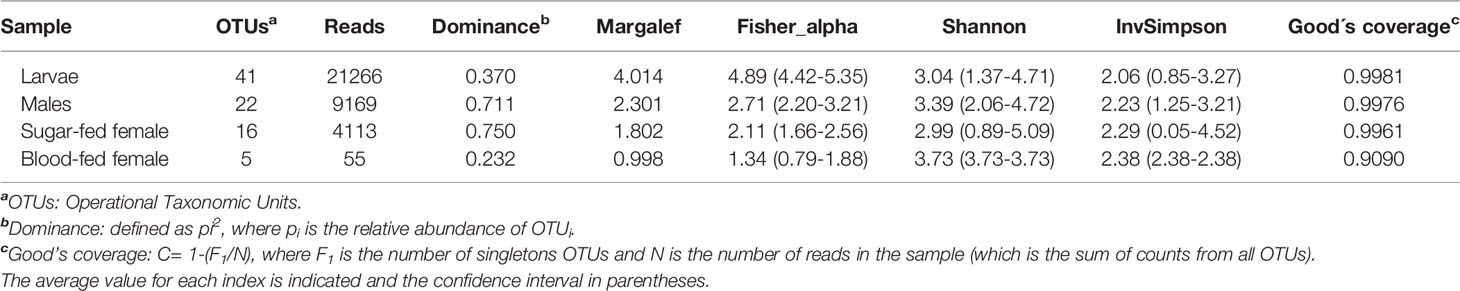
Table 1 Summary of community richness and alpha diversity per Cx. quinquefasciatus instars.
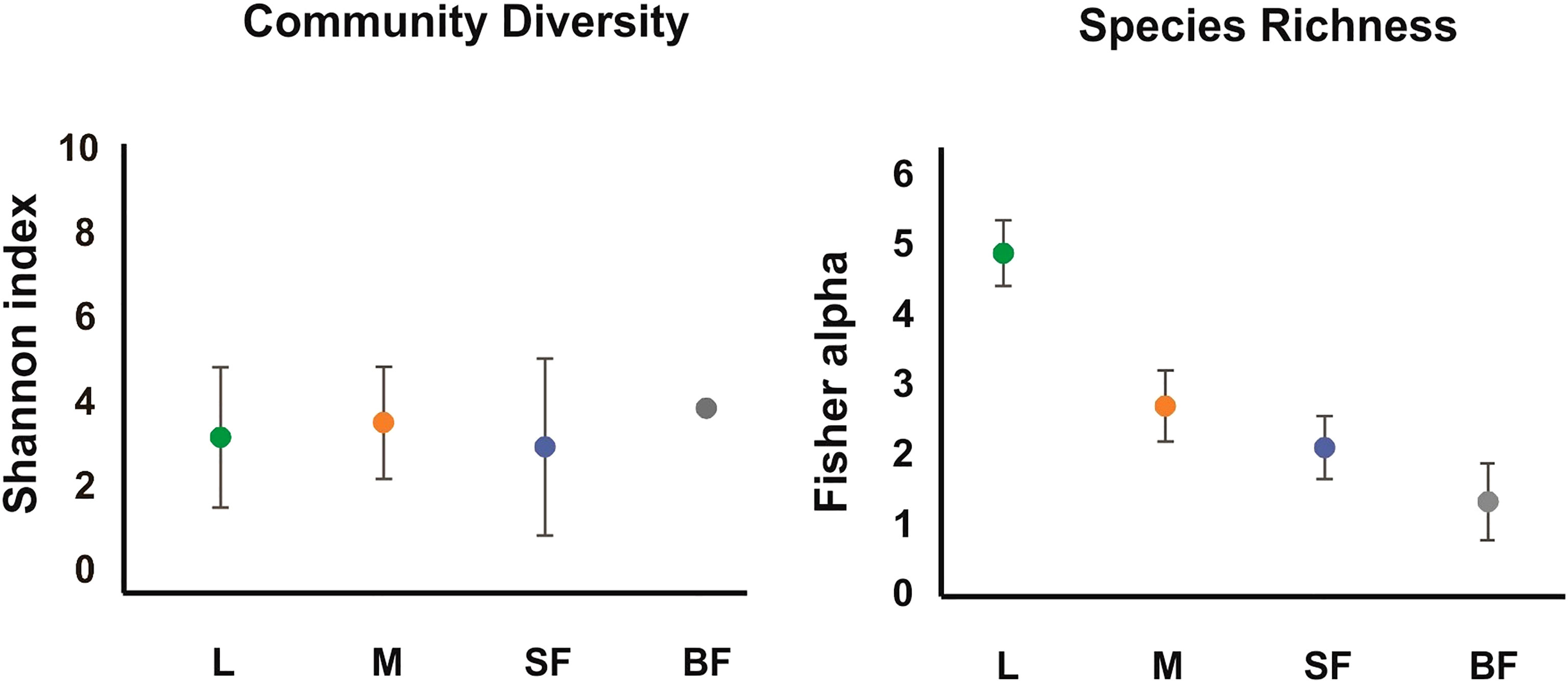
Figure 1 Alpha diversity of the fungal communities along Cx. quinquefasciatus development. The alpha-diversity was estimated with diverse indexes for different instars. The Fisher_alpha, and Shannon indices are represented in the graphs. L, larvae; M, sucrose-fed male; SF, sucrose-fed females; BF, blood-fed females. The error bars represent the 95% confidence interval, obtained with a randomization of 100 runs and sampling with replacement.
On the other hand, to evaluate species richness in the different mosquito stages Margalef and Fisher_alpha indexes were calculated (Table 1 and Figure 1), observing that the larvae present greater richness than the adult stages. Indeed, fungal species richness seems to decrease with mosquito development. 41 fungal OTUs were identified in the larval stage (74.54% of the total OTUs from all samples), in which 27 were exclusively in this stage. 6 and 3 unique fungal OTUs were identified in adult males and females fed on sucrose solution respectively, while 2 were detected only in blood-fed females. Furthermore, although there seem to be no major differences between males and females, a significant decrease in species richness was observed once the adult females were fed on blood.
Fungal Beta Diversity
To analyze the similarity between fungal communities in different mosquito instars and sex, throughout the life cycle of Cx. quinquefasciatus laboratory line, NMDS and PCA were performed. The general comparison allowed us to observe that there are distinct clusters that differentially grouped the fungal communities of larval and adult samples (Figures 2, 3). Moreover, samples from the adult stage present a similar fungal community regardless of whether they are male or female. These observations are supported by a high similarity value of Sorensen index between males and sucrose-fed females (0.63) and lower values for larvae and adults (0.13 - 0.41), blood-fed females and the other groups (0.13 - 0.22) (Supplementary Table 3).
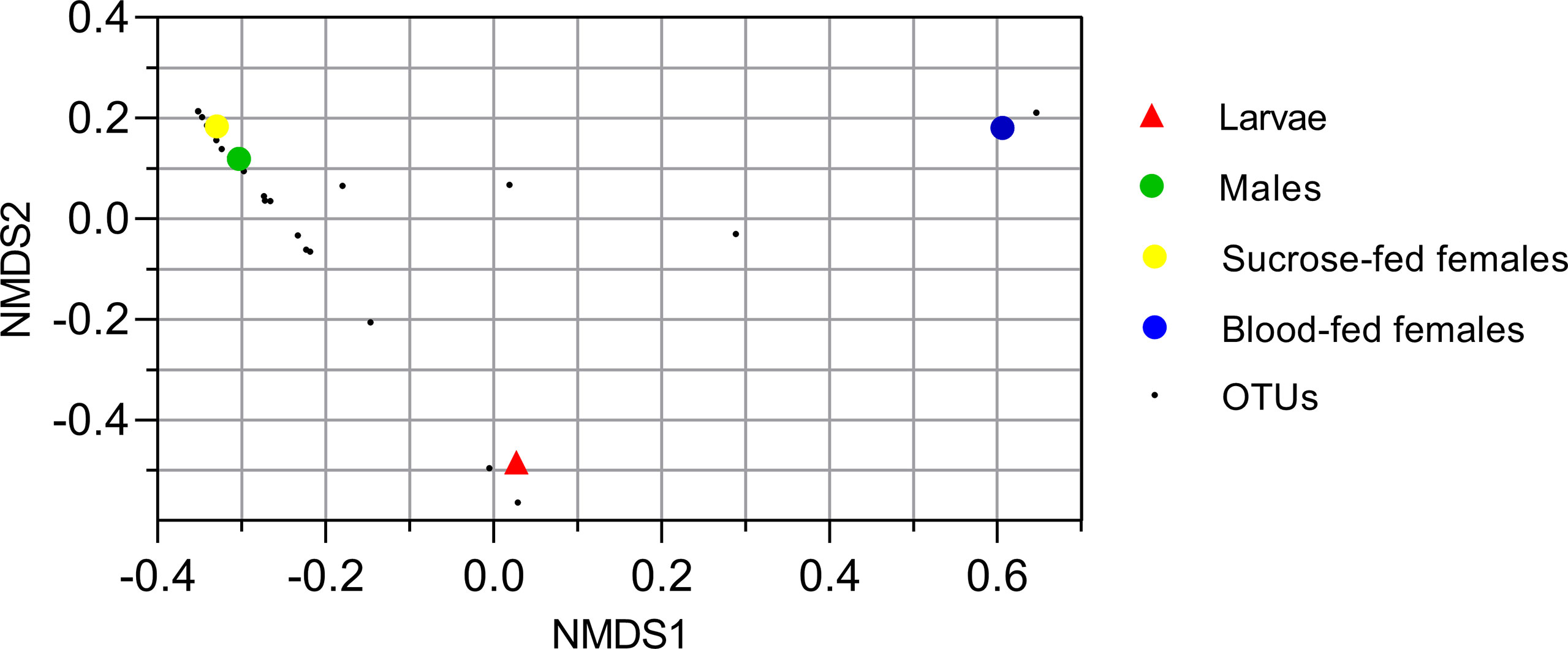
Figure 2 Nonmetric multidimensional scale ordination plot of groups based on Bray-Curtis dissimilarity matrix. Fungal operational taxonomic units (OTUs) are represented with small black points. Larvae, male and sucrose-fed females are in duplicates.
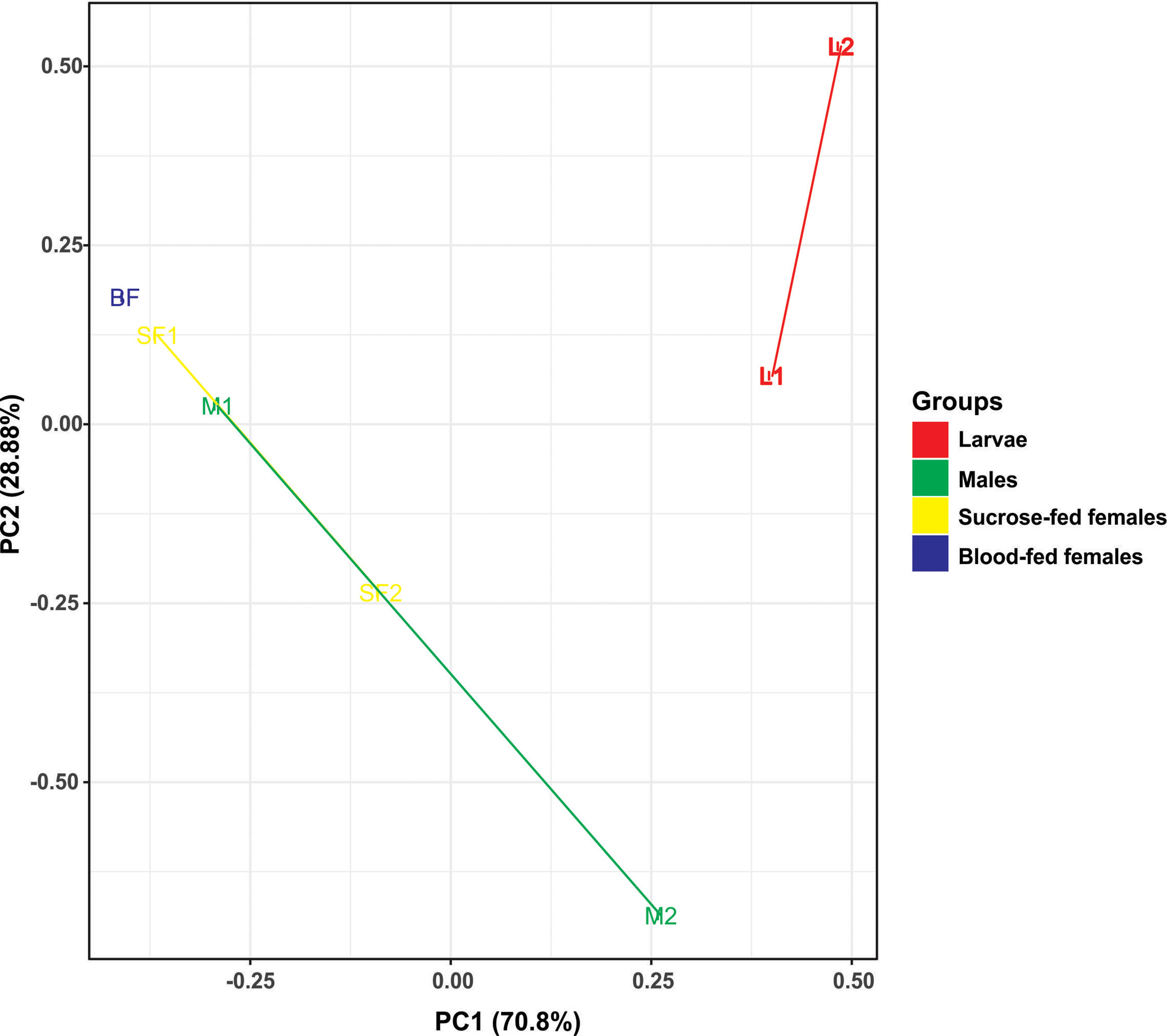
Figure 3 Principal component analysis (PCA) based on the Euclidean distances for fungal communities from samples along the Cx. quinquefasciatus development. There are two distinct groups, adults and larvae, using two principal components (PC1 and PC2).
Fungal Communities Composition in Cx. quinquefasciatus
The composition of the Cx. quinquefasciatus fungal communities at different taxonomic levels was analyzed, assigning the taxonomic nomenclature of each OTU through the UNITE database. The identified OTUs corresponded to 4 phyla, Ascomycota (58%), Basidiomycota (34.5%) Mucoromycota (0.036%) and Morteriellomycota (0.018%), although other 2 OTUs were unidentified. Even though, the taxonomic affiliation of the sequences was able to identify fungal species level, in some cases, only phylum was reached.
At the phylum level, Ascomycota was the most abundant in all stages, representing more than 90% of the sequences in sucrose-fed males and females (Figure 4). In these groups more than 80% of the Ascomycota abundance was due to the presence of Penicillium polonicum (Eurotiomycetes). This fungus was detected in all stages with a relative abundance between 24 to 86% (Table 2). Particularly, it was predominant in sucrose-fed adults (84.10% males and 86.21% females); however, in larvae the relative abundance was 42.8% and 23.64% in blood-fed females. In comparison, the phylum Basidiomycota presented a low percentage in all stages (up to 2.4%), except for blood-fed females in which it was represented by 25.45% of their total fungal sequences, composed of a single OTU, corresponding to the Pucciniomycete Puccinia sorghi, found only in this stage. The remaining sequences, corresponding to Basidiomycota, were identified as subphylum Agaricomycotina, with classes Agaricomycetes and Tremellomycetes, including filamentous fungi (46.9%) and yeasts (53.07%). In particular, yeasts or yeast-like sequences detected in Cx. quinquefasciatus, belong to the sub-phyla Pucciniomycotina and Agaricomycotina (Basidiomycota), Pezizomycotina and Saccharomycotina (Ascomycota).
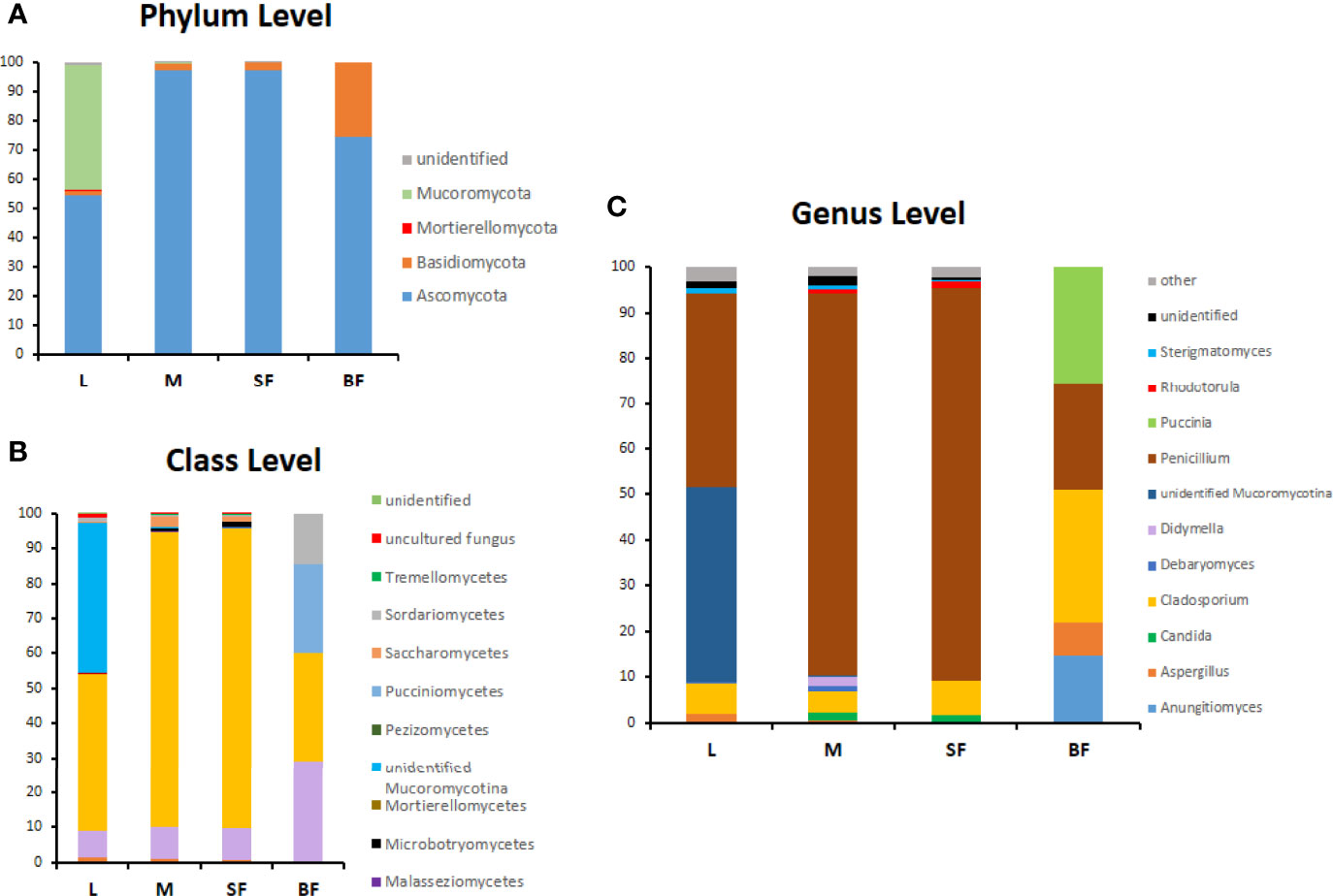
Figure 4 Relative abundance (%) of fungi along Cx. quinquefasciatus development at the phylum (A), class (B) and genus level (C). OTUs with values >1% were grouped under other genera in panel (C).
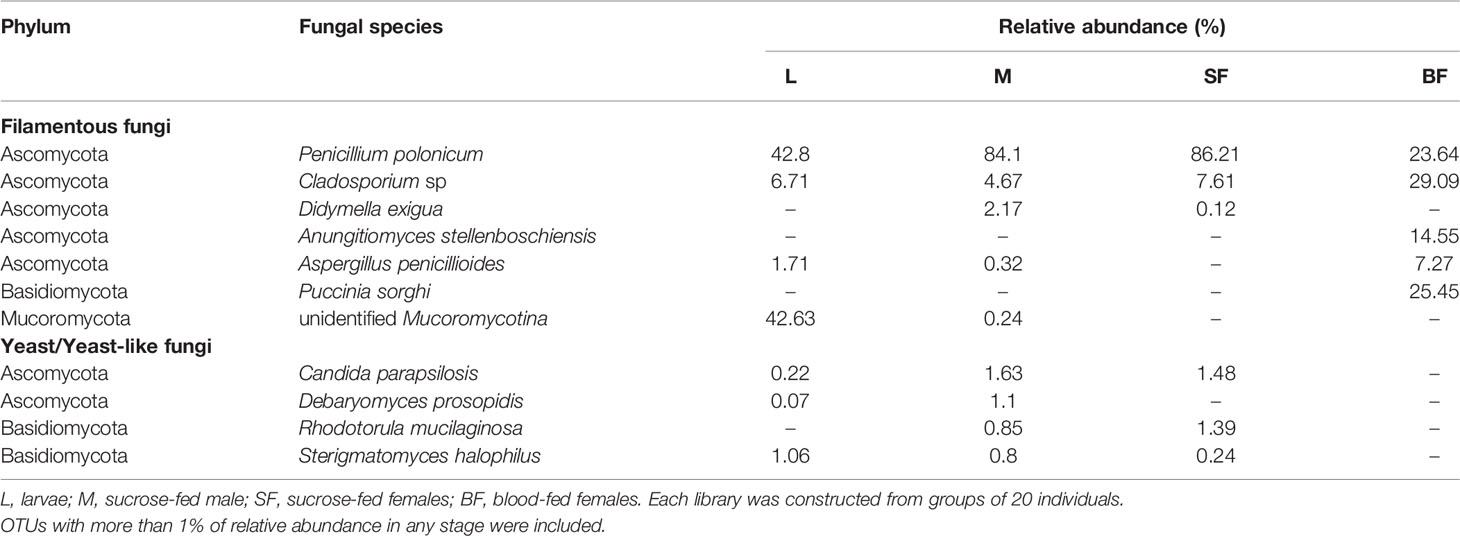
Table 2 Fungal classified OTUs identified throughout Cx. quinquefasciatus development.
The larval stage showed the greatest diversity of fungal species since 41 OTUs were determined, with dominance of Penicillium and Mucoromycotina (approximately 43% abundance each). 7 OTUs (12.7%) are shared by three groups (larvae, males, and sucrose-fed females), belonging to the following Ascomycota orders: Agaricostilbales, Saccharomycetales, Capnodiales, Hypocreales, Pleosporales, Capnodiales, Malasseziales and 1 OTU identified as member of the Aspergillaceae family is shared by larvae, males and blood-fed females. Sucrose- and blood-fed females are the 2 groups that share the minor amount of OTUs, in which Penicillium and Cladosporium were present in both of them. Sucrose-fed females and males share 3 OTUs of Didymella, Rhodotorula and Cutaneotrichosporon yeast genera; while larvae and males from Mucoromycotina, Debaryomyces and Aspergillus.
Finally, the blood-fed females present a lower richness but with an even distribution of the OTUs (Penicillium, Cladosporium and Puccinia which were present in a proportion of nearly 25% each one), sharing fewer OTUs with the other groups. OTUs shared in all mosquito stages are shown in Figure 5 and Supplementary Table 2.
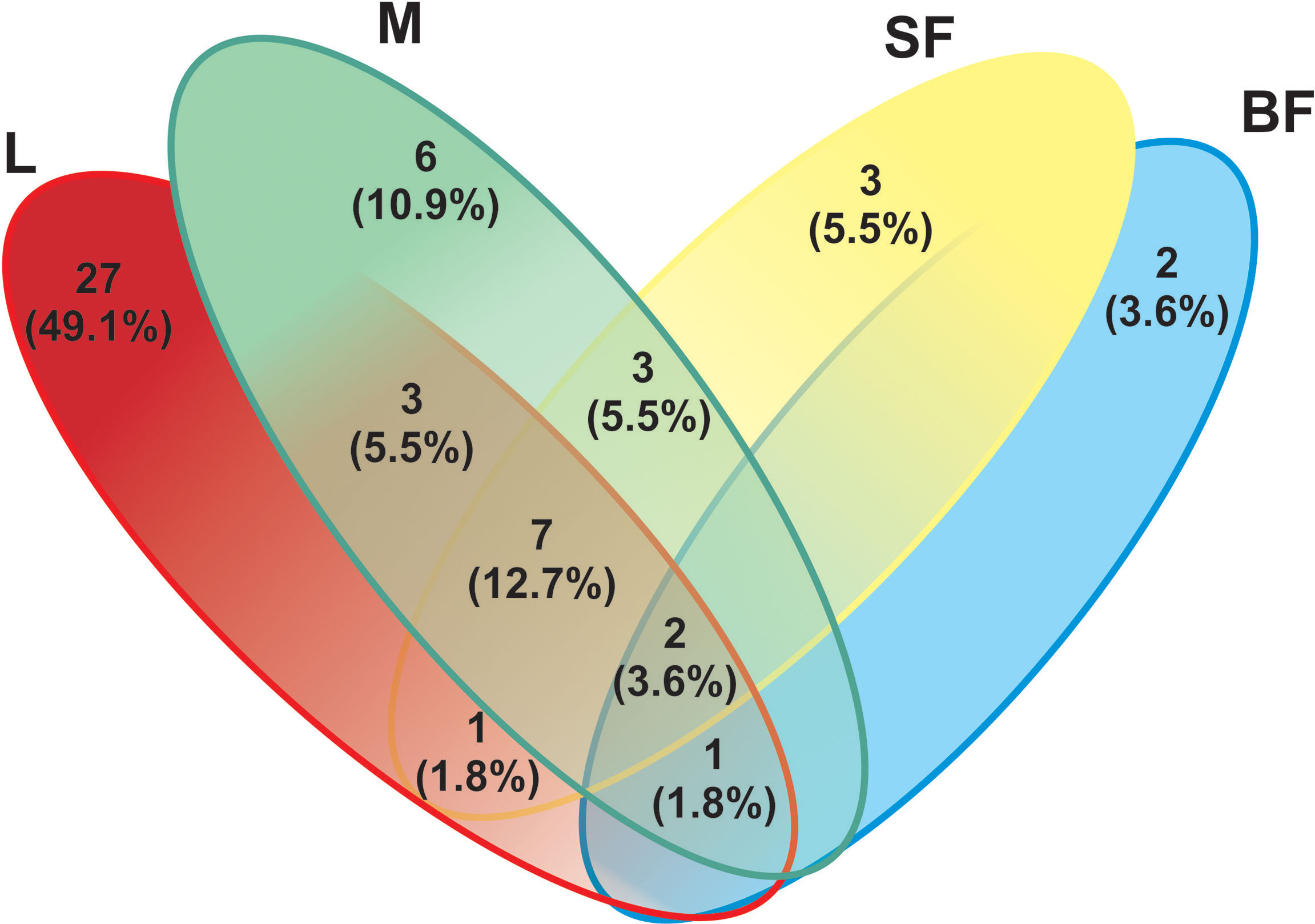
Figure 5 Venn-diagram depicts the number of fungal ITS Operational Taxonomic Units (OTUs) overlap and non-overlap between Cx. quinquefasciatus instars. A total of 55 fungal OTUs (based on the sequencing of the ITS-2 regions) were detected. L, larvae; M, male; SF, sucrose-fed females; and BF, blood-fed females.
Discussion
Research on the mosquito microbiome has increased significantly during the last years, highlighting its role in the biology of mosquitoes and its potential as agents for the development of control strategies for diseases transmitted by them (Dada et al., 2021). In this context, species of microorganisms associated with the mosquito holobiont that affect its nutrition, development and susceptibility to pathogens, and its vectorial competence as well, have been described through the use of culture-dependent and independent techniques (Dennison et al., 2014; Duguma et al., 2015; Guégan et al., 2018). In particular, the main efforts have been made on bacteria, while few studies have described the whole fungal communities associated with these insects. In this work, the fungal composition of Cx. quinquefasciatus mosquito larvae and adults, including female blood-fed, reared in laboratory conditions, were characterized through a metagenomic approach. The highest fungal richness was observed in larval stages, such as in mosquitoes reared in natural sites, even though our population was bred under controlled laboratory conditions. This result agrees with Zouache et al. (2022), who suggested that the mosquito microbiota could be acquired during their feeding habit of filtering and grazing. In particular, the diversity and composition of mosquito mycobiota has been determined, at least partially, by the diversity and composition of microorganisms in their habitats (Thongsripong et al., 2017), as it has been observed in Ae. albopictus larval guts (Tawidian et al., 2021). Decrease in fungal diversity between Cx. quinquefasciatus larvae and all adult samples could be explained by the effects of the metamorphosis process, in which mosquito larvae suffer drastic morphological changes that reduce their microbial abundance, especially by the egestion of the last larval food bolus and then of the midgut meconium in newly emerged adults (Moll et al., 2001) (Table 1).
It has been reported that fungal microbiome communities of several mosquito species are composed mainly by the phylum Ascomycota (especially Pezizomycotina), and Basidiomycota in a lower proportion (Muturi et al., 2016; Thongsripong et al., 2017; Luis et al., 2019; Zouache et al., 2022). Thus, in our work and according to these studies, diversity, and phyla distribution observed was dominated by Ascomycota fungi. Among the fungal OTUs, 2 of them were widespread in almost all individuals. The most abundant species was P. polonicum, a common filamentous saprophytic fungus, which was found in all mosquito instars, mostly in adults as male and sucrose-fed females (Table 2). This fungal genus was found associated with mosquitoes, in many studies including culture dependent and independent approaches (Pereira et al., 2009; Muturi et al., 2016; Luis et al., 2019; Zouache et al., 2022). Although this saprophyte could be maintained throughout the mosquito development, the role in its life cycle is unknown. It has been pointed out that this fungus produces several metabolites that can be used in insect control (Ragavendran et al., 2019). In this way a Penicillium citrinum strain showed high mortality rates against Cx. quinquefasciatus larvae, due partially to patulin, a metabolite secreted by this fungus (Maketon et al., 2014). Taken together, these observations suggest that P. polonicum as other related saprobes could have a role that is still unknown in mosquitoes and may be a candidate for future studies in Cx. quinquefasciatus control strategies.
Additionally, we found Cladosporium sp., another filamentous ascomycete in all mosquito stages. This genus was found in Cx. quinquefasciatus populations collected from natural habitats, and in other important mosquito vectors like Cx. pipiens and Ae. albopictus as well, suggesting that its presence in adult females may be due to its occurrence in mosquito environments (Chandler et al., 2015; Thongsripong et al., 2017; Luis et al., 2019). Cladosporium is one of the commonest genera in indoor environments, and under certain conditions some species become predominant (Bensch et al., 2018). Maybe Cx. quinquefasciatus could have acquired it from the natural environment, and later passed on to its progeny, keeping it throughout development and in its breeding site in our laboratory conditions.
Another identified filamentous fungi group belonged to the phylum Mucoromycota and subphylum Morteriellomycotina which was present in a low percentage (Table 2). It was a dominant on larval stage (approximately 43% abundance). It was not possible to obtain deeper taxonomic information about the OTU of this group. Thus, these fungi are reported as a diverse group but for the majority of taxa, their ecological role and their geographic distribution remain unknown (Walther et al., 2019).
Yeast or yeast-like Ascomycetes and Basidiomycetes were also identified as an important part of the mycobiome in our mosquito population, although in low total relative abundance (less than 5%). This group included species like Candida parapsilosis, Sterigmatomyces halophilus and Debaryomyces prosopidis in larvae and sucrose-fed adults, while Rhodotorula mucilaginosa was only found in sucrose-fed adults (Table 2). Candida sp. was reported as part of the “core microbiome” in Ae. albopictus (Luis et al., 2019) and C. parapsilosis stably associated with several mosquito species, including Cx. quinquefasciatus (Bozic et al., 2017). Also, Steyn et al. (2016) were able to isolate, through culturable methods, not only Candida spp. but also Rhodotorula spp. and Hannaella sp. in Culex mosquitoes, indicating that adults harbored much less yeasts than larvae, suggesting a diminished transmission to the mature stage. This data is consistent with our results, being this the first report of S. halophilus and D. prosopidis in mosquitoes.
In nature, fungal colonization of the mosquito body probably may occur during the contact and ingestion of plant sugar products. Particularly, in adult mosquitoes fed on nectar and captured in the field, many yeast genera, also present on flowers, were detected (Luis et al., 2019). In this work, laboratory-raised adults were fed on sucrose solution, but diluted in non-sterile dechlorinated water. Novak Babič et al. (2016) studying tap water fungal communities identified different yeast and yeast-like as common species in drinking water. Among them R. mucilaginosa, C. parapsilosis and Aureobasidium sp. occurred in Cx. quinquefasciatus adults, probably explaining the presence of certain fungal species in mature stages in this work. Interestingly, R. mucilaginosa was only found in sugar-fed males and females. Moreover, some filamentous fungi like Cladosporium and Aspergillus, especially metabolically active in males, could have a role in fructose digestion, as analyzed in Ae. albopictus adult (Guégan et al., 2020b). The presence of the same genera in our laboratory mosquito line, indicates that adult feeding habits could be contributing not only to fungal recolonization after adult emergence, but also to incorporation of fungal symbionts involved in nutrition.
Cx. quinquefasciatus females storage nutrients after blood ingestion, increase their reproductive fitness, since blood digestion confers lipids associated with longevity and proteins provisioning yolk for egg production. The blood meal produces changes in gut physiology, modifying the microbiota relative abundance, well studied in bacterial communities (Wang et al., 2011; Telang and Skinner, 2019). However, there are few reports about the fungal diversity changes in females, as seen for the yeast Meyerozyma, where its proliferation was favored in two Aedes species after blood ingestion (Muturi et al., 2016). In this study we observed a strong change in OTUs dominance in blood-fed females, compared to the sucrose-fed adults: P. polonicum abundance decreased and Cladosporium became dominant, besides the fungal community was reduced to 5 OTUs with an equitative distribution between species.
Fungal communities in Cx. quinquefasciatus presented significant specific signatures, showing differences in composition across their life stage and among reproductive states. Indeed, beta diversity analyses showed a pattern of sample-related clustering composition in agreement with the specific lifestyle of each developmental stage, indicating the presence of sample-type related mycobiota. Species composing the “core mycobiota” in males and females differ, almost completely, from the larval microbiota, suggesting that striking differences in larval diet, sucrose-based adults, and blood-fed females, can generate changes in the intestinal microbial diversity and composition.
In summary, our results provide information on the fungal composition of mosquitoes reared exclusively under controlled laboratory conditions. We suggest that the described mycobiota may be essential for mosquito survival and could have important implications in their biology and pathogen transmission. Species from the mosquito “core mycobiota” could be considered as potential targets and could open the possibility to further experimental studies which exploit them as potential microorganisms in the development of new biotechnological strategies for the mosquito vector-borne diseases control.
Data Availability Statement
The data presented in the study are deposited in the NCBI repository, accession number PRJNA828597.
Author Contributions
GF, RL, VC, and CB designed the research plan. GF, RL, and CC performed the bioinformatics analysis. VC and CB acquired funding and supervised the work. All authors contributed to result analysis, wrote and edited the manuscript, read, and approved the submitted version.
Funding
This study was supported by Grants of the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), PUE 2017-0101 (CB) and PIP 2021-2023 1012 (VC), and Universidad Nacional de Mar del Plata (15/E883 EXA925/19) (CB).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We especially thank Prof. Ana Tassi (former Professor of English Grammar, Universidad Nacional de Mar del Plata) for proof-editing of the draft manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffunb.2022.918052/full#supplementary-material
Supplementary Figure 1 | Algorithm representing the analysis performed in this work. For the fungi molecular identification amplicon sequencing based on Internal Transcribed Spacer (ITS) were used. After sequence processing and analysis, the resulting ITS sequences were compared against a known database and the fungal species were identified. In this work we used several tools according with Gao et al. (2021) following the main steps: quality control (grey box) and data preprocessing (yellow box), clustering of reads in OTUs (blue box) and taxonomic assignment (green box), community characterization (orange box). In the present work we used genomic DNA of larvae, males, females only fed on sucrose solution and females fed on mouse blood for fungal ITS2 amplicon sequencing, with an Illumina Miseq 300 bp paired-end (PE) platform. The databases, the programs and their main parameters used in each step are indicated.
Supplementary Figure 2 | Rarefaction curves described for observed OTUs metric among all mosquito groups: L, larvae; M, sucrose-fed male; SF, sucrose-fed females; BF, blood-fed females. The rarefied number of operational taxonomic units (OTUs, with 97% sequence similarity cut-off value) is plotted against the number of reads sampled.
Supplementary Data Sheet 1 | Krona plots of Cx. quinquefasciatus fungal holobiont identified in each development instar. L, larvae; M, sucrose-fed male; SF, sucrose-fed females; BF, blood-fed females. The results are illustrated in an interactive zooming and multi-layered fungal map obtained by Krona (html format), showing the mean taxonomic distribution and relative abundance of taxa.
Supplementary Table 2 | Reads assigned as fungal OTUs at 97% identity from all mosquito stages: L, larvae; M, sucrose-fed male; SF, sucrose-fed females; BF, blood-fed females.
References
Afgan E., Baker D., Batut B., Van Den Beek M., Bouvier D., Čech M., et al. (2018). The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 46, W537–W544. doi: 10.1093/nar/gky379
Becker N., Petrić D., Zgomba M., Boase C., Madon M. B., Dahl C., et al. (2020). Mosquitoes: Identification, Ecology and Control (Switzerland: Springer International Publishing).
Bellemain E., Carlsen T., Brochmann C., Coissac E., Taberlet P., Kauserud H. (2010). ITS as an Environmental DNA Barcode for Fungi: An in Silico Approach Reveals Potential PCR Biases. BMC Microbiol. 10, 1–9. doi: 10.1186/1471-2180-10-189
Bensch K., Groenewald J. Z., Meijer M., Dijksterhuis J., Jurjević Ž., Andersen B., et al. (2018). Cladosporium Species in Indoor Environments. Stud. Mycol. 89, 177–301. doi: 10.1016/j.simyco.2018.03.002
Bian G., Joshi D., Dong Y., Lu P., Zhou G., Pan X., et al. (2013). Wolbachia Invades Anopheles Stephensi Populations and Induces Refractoriness to Plasmodium Infection. Science 340, 748–751. doi: 10.1126/science.1236192
Bozic J., Capone A., Pediconi D., Mensah P., Cappelli A., Valzano M., et al. (2017). Mosquitoes can Harbour Yeasts of Clinical Significance and Contribute to Their Environmental Dissemination. Environ. Microbiol. Rep. 9, 642–648. doi: 10.1111/1758-2229.12569
Chandler J. A., Liu R. M., Bennett S. N. (2015). RNA Shotgun Metagenomic Sequencing of Northern California (USA) Mosquitoes Uncovers Viruses, Bacteria, and Fungi. Front. Microbiol. 06. doi: 10.3389/fmicb.2015.00185
Coon K. L., Brown M. R., Strand M. R. (2016). Mosquitoes Host Communities of Bacteria That are Essential for Development But Vary Greatly Between Local Habitats. Mol. Ecol. 25, 5806–5826. doi: 10.1111/mec.13877
Dada N., Jupatanakul N., Minard G., Short S. M., Akorli J., Villegas L. M. (2021). Considerations for Mosquito Microbiome Research From the Mosquito Microbiome Consortium. Microbiome 9, 36. doi: 10.1186/s40168-020-00987-7
Dennison N. J., Jupatanakul N., Dimopoulos G. (2014). The Mosquito Microbiota Influences Vector Competence for Human Pathogens. Curr. Opin. Insect Sci. 3, 6–13. doi: 10.1016/j.cois.2014.07.004
Díaz-Nieto L. M., Gil M. F., Lazarte J. N., Perotti M. A., Berón C. M. (2021). Culex Quinquefasciatus Carrying Wolbachia Is Less Susceptible to Entomopathogenic Bacteria. Sci. Rep. 11, 1–9. doi: 10.1038/s41598-020-80034-5
Dong Y., Manfredini F., Dimopoulos G. (2009). Implication of the Mosquito Midgut Microbiota in the Defense Against Malaria Parasites. PLoS Pathog. 5, e1000423. doi: 10.1371/journal.ppat.1000423
Duguma D., Hall M. W., Rugman-Jones P., Stouthamer R., Terenius O., Neufeld J. D., et al. (2015). Developmental Succession of the Microbiome of Culex Mosquitoes. BMC Microbiol. 15, 1–13. doi: 10.1186/s12866-015-0475-8
Duron O., Labbé P., Berticat C., Rousset F., Guillot S., Raymond M., et al. (2006). High Wolbachia Density Correlates With Cost of Infection for Insecticide Resistant Culex Pipiens Mosquitoes. Evolution 60, 303–314. doi: 10.1111/j.0014-3820.2006.tb01108.x
Edgar R. C. (2013). UPARSE: Highly Accurate OTU Sequences From Microbial Amplicon Reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Gao B., Chi L., Zhu Y., Shi X., Tu P., Li B., et al. (2021). An Introduction to Next Generation Sequencing Bioinformatic Analysis in Gut Microbiome Studies. Biomolecules 11, 530. doi: 10.3390/biom11040530
Gil M. F., Fassolari M., Battaglia M. E., Berón C. M. (2021). Culex Quinquefasciatus Larvae Development Arrested When Fed on Neochloris Aquatica. PLoS Negl. Trop. Dis. 15, e0009988. doi: 10.1038/s41598-020-80034-5
Guégan M., Martin E., Moro C. V. (2020a). Comparative Analysis of the Bacterial and Fungal Communities in the Gut and the Crop of Aedes Albopictus Mosquitoes: A Preliminary Study. Pathogens 9, 628. doi: 10.3390/pathogens9080628
Guégan M., Van V. T., Martin E., Minard G., Tran F.-H., Fel B., et al. (2020b). Who is Eating Fructose Within the Aedes Albopictus Gut Microbiota? Environ. Microbiol. 22, 1193–1206. doi: 10.1111/1462-2920.14915
Guégan M., Zouache K., Démichel C., Minard G., Tran Van V., Potier P., et al. (2018). The Mosquito Holobiont: Fresh Insight Into Mosquito-Microbiota Interactions. Microbiome 6, 49. doi: 10.1186/s40168-018-0435-2
Guerrero R., Margulis L., Berlanga M. (2013). Symbiogenesis: The Holobiont as a Unit of Evolution. Int. Microbiol. 16, 133–143. doi: 10.2436/20.1501.01.188
Gusmão D. S., Santos A. V., Marini D. C., Bacci M., Berbert-Molina M. A., Lemos F. J. A. (2010). Culture-Dependent and Culture-Independent Characterization of Microorganisms Associated With Aedes Aegypti (Diptera: Culicidae) (L.) and Dynamics of Bacterial Colonization in the Midgut. Acta Trop. 115, 275–281. doi: 10.1016/j.actatropica.2010.04.011
Harvey-Samuel T., Ant T., Sutton J., Niebuhr C. N., Asigau S., Parker P., et al. (2021). Culex Quinquefasciatus: Status as a Threat to Island Avifauna and Options for Genetic Control. CABI Agric. Biosci. 2, 1–21. doi: 10.1186/s43170-021-00030-1
Hoffmann A. A., Montgomery B. L., Popovici J., Iturbe-Ormaetxe I., Johnson P. H., Muzzi F., et al. (2011). Successful Establishment of Wolbachia in Aedes Populations to Suppress Dengue Transmission. Nature 476, 454–457. doi: 10.1038/nature10356
Huang W., Wang S., Jacobs-Lorena M. (2020). Use of Microbiota to Fight Mosquito-Borne Disease. Front. Genet. 11. doi: 10.3389/fgene.2020.00196
Luis P., Vallon L., Tran F.-H., Hugoni M., Tran-Van V., Mavingui P., et al. (2019). Aedes Albopictus Mosquitoes Host a Locally Structured Mycobiota With Evidence of Reduced Fungal Diversity in Invasive Populations. Fungal Ecol. 39, 257–266. doi: 10.1016/j.funeco.2019.02.004
Maketon M., Amnuaykanjanasin A., Kaysorngup A. (2014). A Rapid Knockdown Effect of Penicillium Citrinum for Control of the Mosquito Culex Quinquefasciatus in Thailand. World J. Microbiol. Biotechnol. 30, 727–736. doi: 10.1007/s11274-013-1500-4
Malassigné S., Valiente Moro C., Luis P. (2020). Mosquito Mycobiota: An Overview of non-Entomopathogenic Fungal Interactions. Pathogens 9, 564. doi: 10.3390/pathogens9070564
Moll R. M., Romoser W. S., Modrakowski M. C., Moncayo A. C., Lerdthusnee K. (2001). Meconial Peritrophic Membranes and the Fate of Midgut Bacteria During Mosquito (Diptera: Culicidae) Metamorphosis. J. Med. Entomol. 38, 29–32. doi: 10.1603/0022-2585-38.1.29
Muturi E. J., Bara J. J., Rooney A. P., Hansen A. K. (2016). Midgut Fungal and Bacterial Microbiota of Aedes Triseriatus and Aedes Japonicus Shift in Response to La Crosse Virus Infection. Mol. Ecol. 25, 4075–4090. doi: 10.1111/mec.13741
Nilsson R. H., Larsson K.-H., Taylor A. F. S., Bengtsson-Palme J., Jeppesen T. S., Schigel D., et al. (2019). The UNITE Database for Molecular Identification of Fungi: Handling Dark Taxa and Parallel Taxonomic Classifications. Nucleic Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Novak Babič M., Zalar P., Ženko B., Džeroski S., Gunde-Cimerman N. (2016). Yeasts and Yeast-Like Fungi in Tap Water and Groundwater, and Their Transmission to Household Appliances. Fungal Ecol. 20, 30–39. doi: 10.1016/j.funeco.2015.10.001
Oksanen J., Blanchet G. F., Friendly M., Kindt R., Legendre P., McGlinn D., et al. (2020) Vegan: Community Ecology Package (Version 2.5-6, R Package 2019). Available at: http://CRAN.R-project.org/package=vegan.
Ondov B. D., Bergman N. H., Phillippy A. M. (2011). Interactive Metagenomic Visualization in a Web Browser. BMC Bioinf. 12, 385. doi: 10.1186/1471-2105-12-385
Pereira E., da S., de M M. I., Ferreira-Keppler R. L., Hamada N., Alencar Y. B. (2009). Filamentous Fungi Associated With Mosquito Larvae (Diptera: Culicidae) in Municipalities of the Brazilian Amazon. Neotrop Entomol. 38, 352–359. doi: 10.1590/S1519-566X2009000300009
Ragavendran C., Manigandan V., Kamaraj C., Balasubramani G., Prakash J. S., Perumal P., et al. (2019). Larvicidal, Histopathological, Antibacterial Activity of Indigenous Fungus Penicillium Sp. Against Aedes Aegypti L and Culex Quinquefasciatus (Say) (Diptera: Culicidae) and its Acetylcholinesterase Inhibition and Toxicity Assessment of Zebrafish (Danio Rerio). Front. Microbiol. 10. doi: 10.3389/fmicb.2019.00427
RC Team (2021). R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: http://www.R-project.org/.
Ricci I., Mosca M., Valzano M., Damiani C., Scuppa P., Rossi P., et al. (2011). Different Mosquito Species Host Wickerhamomyces Anomalus (Pichia Anomala): Perspectives on Vector-Borne Diseases Symbiotic Control. Antonie Van Leeuwenhoek 99, 43–50. doi: 10.1007/s10482-010-9532-3
Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Singh U. S., Das A. (2020). Vector-Borne Diseases: Mosquito Holobiont and Novel Methods for Vector Control. Asian Pac. J. Trop. Biomed. 10, 341–342. doi: 10.4103/2221-1691.287160
Steyn A., Roets F., Botha A. (2016). Yeasts Associated With Culex Pipiens and Culex Theileri Mosquito Larvae and the Effect of Selected Yeast Strains on the Ontogeny of Culex Pipiens. Microb. Ecol. 71, 747–760. doi: 10.1007/s00248-015-0709-1
Tawidian P., Coon K. L., Jumpponen A., Cohnstaedt L. W., Michel K. (2021). Host-Environment Interplay Shapes Fungal Diversity in Mosquitoes. mSphere 6, e00646-21. doi: 10.1128/mSphere.00646-21
Telang A., Skinner J. (2019). Effects of Host Blood Meal Source on Reproductive Output, Nutrient Reserves and Gut Microbiome of West Nile Virus Vector Culex Quinquefasciatus. J. Insect Physiol. 114, 15–22. doi: 10.1016/j.jinsphys.2019.02.001
Thongsripong P., Chandler J. A., Green A. B., Kittayapong P., Wilcox B. A., Kapan D. D., et al. (2017). Mosquito Vector-Associated Microbiota: Metabarcoding Bacteria and Eukaryotic Symbionts Across Habitat Types in Thailand Endemic for Dengue and Other Arthropod-Borne Diseases. Ecol. Evol. 8, 1352–1368. doi: 10.1002/ece3.3676
Vinogradova E. B. (2000). Culex Pipiens Pipiens Mosquitoes: Taxonomy, Distribution, Ecology, Physiology, Genetics, Applied Importance and Control (Sofia, Bulgaria: Pensoft Publishers).
Walther G., Wagner L., Kurzai O. (2019). Updates on the Taxonomy of Mucorales With an Emphasis on Clinically Important Taxa. J. Fungi 5, 106. doi: 10.3390/jof5040106
Wang Y., Iii T. M. G., Kukutla P., Yan G., Xu J. (2011). Dynamic Gut Microbiome Across Life History of the Malaria Mosquito Anopheles Gambiae in Kenya. PLoS One 6, e24767. doi: 10.1371/journal.pone.0024767
WHO (2020) Vector-Borne Diseases. Available at: https://www.who.int/news-room/fact-sheets/detail/vector-borne-diseases (Accessed April 4, 2022).
Yun J.-H., Roh S. W., Whon T. W., Jung M.-J., Kim M.-S., Park D.-S., et al. (2014). Insect Gut Bacterial Diversity Determined by Environmental Habitat, Diet, Developmental Stage, and Phylogeny of Host. Appl. Environ. Microbiol. 80, 5254–5264. doi: 10.1128/AEM.01226-14
Keywords: mosquito, Culex quinquefasciatus, mycobiota, metagenomics, ITS2
Citation: Flores GAM, Lopez RP, Cerrudo CS, Consolo VF and Berón CM (2022) Culex quinquefasciatus Holobiont: A Fungal Metagenomic Approach. Front. Fungal Biol. 3:918052. doi: 10.3389/ffunb.2022.918052
Received: 12 April 2022; Accepted: 24 June 2022;
Published: 02 August 2022.
Edited by:
Nicolas Pedrini, National University of La Plata, ArgentinaReviewed by:
Sebastian Ulrich, Ludwig Maximilian University of Munich, GermanyJose Luis Ramirez, United States Department of Agriculture (USDA), United States
Copyright © 2022 Flores, Lopez, Cerrudo, Consolo and Berón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: V. Fabiana Consolo, faconsolo@inbiotec-conicet.gob.ar; Corina M. Berón, corina.beron@inbiotec-conicet.gob.ar