 Ning Zhao
Ning Zhao Dan Li
Dan Li Xi Lin
Xi Lin Shuang-Lin Chen
Shuang-Lin Chen- College of Life Sciences, Nanjing Normal University, Nanjing, China
Shiraia bambusicola has long been used as a traditional Chinese medicine and its major medicinal active metabolite is hypocrellin, which exhibits outstanding antiviral and antitumor properties. Here we report the 32 Mb draft genome sequence of S. bambusicola S4201, encoding 11,332 predicted genes. The genome of S. bambusicola is enriched in carbohydrate-active enzymes (CAZy) and pathogenesis-related genes. The phylogenetic tree of S. bambusicola S4201 and nine other sequenced species was constructed and its taxonomic status was supported (Pleosporales, Dothideomycetes). The genome contains a rich set of secondary metabolite biosynthetic gene clusters, suggesting that strain S4201 has a remarkable capacity to produce secondary metabolites. Overexpression of the zinc finger transcription factor zftf, which is involved in hypocrellin A (HA) biosynthesis, increases HA production when compared with wild type. In addition, a new putative HA biosynthetic pathway is proposed. These results provide a framework to study the mechanisms of infection in bamboo and to understand the phylogenetic relationships of S. bambusicola S4201. At the same time, knowledge of the genome sequence may potentially solve the puzzle of HA biosynthesis and lead to the discovery of novel genes and secondary metabolites of importance in medicine and agriculture.
Introduction
Shiraia bambusicola, belonging to the genus Shiraia and the phylum Ascomycota, is an important pathogenic and parasitic fungus that grows on the twigs of bamboos. It adversely affects the growth of bamboo and causes significant economic losses every year. S. bambusicola is widely distributed in many provinces of southern China and Japan (Ali and Olivo, 2002; Morakotkarn et al., 2007). The fruiting body of S. bambusicola has been used as a traditional Chinese medicine for the treatment of sciatica, pertussis, tracheitis, and rheumatic arthritis (Zhao et al., 2016). On the other hand, S. bambusicola is known as the main hyprocrellin-producing species.
The hyprocrellin family includes hypocrellin A (HA), hypocrellin B (HB), hypocrellin C (HC), and hypocrellin D (HD), which are isolated from the parasitic fungi S. bambusicola (Qi et al., 2014) and Hypocrella bambusae (Gu et al., 2005). Resembling many other natural perylenequinones (Figure 1), such as cercosporin and elsinochrome, the hypocrellin family is characterized by a helical chiral pentacyclic conjugated core structure combined with C7, C7′-substitutions, and possessing centrochiral stereochemistry (O’Brien et al., 2010). Because of its high potential pharmacological value, HA has attracted much attention. It has been shown to possess antibacterial, antiviral, antitumor, and anti-inflammatory bioactivities (Guo et al., 2017; Lin et al., 2017). As a photosensitizer, it can be activated under light irradiation and can produce reactive oxygen species (ROS), which can destroy DNA, proteins, and lipids (Robertson et al., 2009). Many researchers are studying the biological characteristics and fermentation production processes of S. bambusicola, as well as the applicability of photodynamic therapy of HA. Although many pharmacological effects of HA are being investigated, HA biosynthetic genes and pathways are still not well understood.
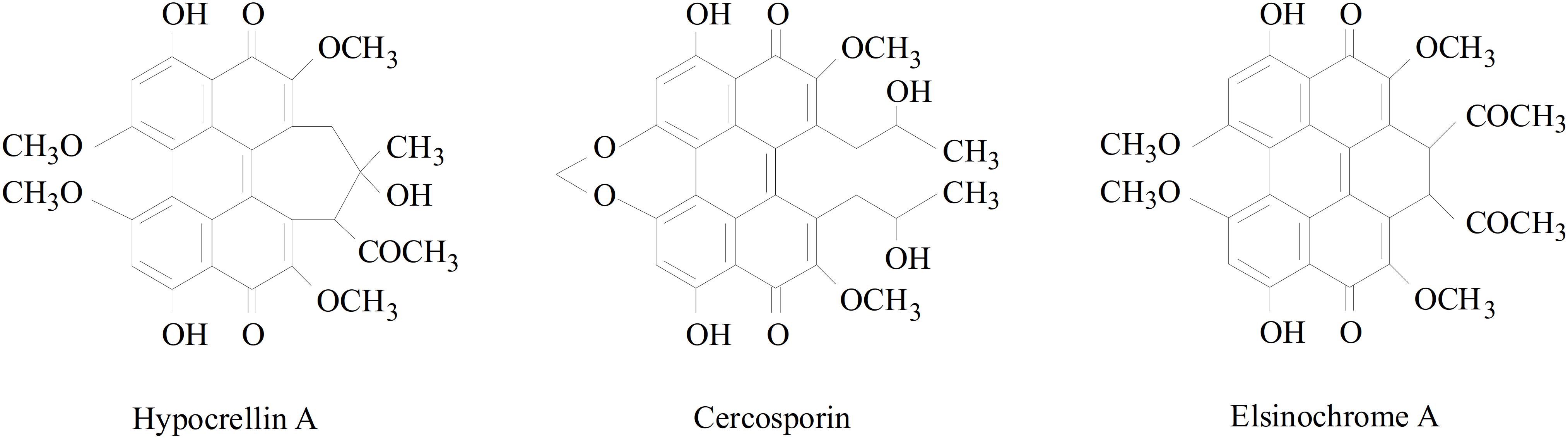
Figure 1. Structures of perylenequinones produced by plant pathogenic fungi.
Fungi have specialized in using plant biomass as a carbon source by producing enzymes that degrade cell wall polysaccharides into metabolizable sugars for nutrition (Rytioja et al., 2014; Han et al., 2016). Carbohydrate-active enzymes (CAZy) are enzymes involved in the synthesis, metabolism, modification, and transport of carbohydrates. It is believed that the number of CAZymes in a fungal species correlates with its life strategies and the nutritional availability (Zhao et al., 2013). The Pathogen–Host Interaction database (PHI-base) contains expertly curated molecular and biological information on genes proven to affect the outcome of pathogen-host interactions. Cytochrome P450s (CYPs) are involved in the synthesis of toxins and pathogenesis (Nelson, 2011). The S. bambusicola S4201 genome data were entered in the databases mentioned above and we found some genes in S. bambusicola that may be involved in pathogenesis, providing a foundation for future studies of its lifestyle.
The taxonomic status of S. bambusicola has been reclassified several times over the past one hundred years. The genus Shiraia was initially classified under Nectriaceae, Hypocreales, and Pyrenomycetes, but was later transferred to the Hypocreaceae family based on the larger fleshy stroma. This taxonomic treatment was popular for several decades, but then Shiraia was transferred to the order Pleosporales based on its bitunicate, as opposed to unitunicate asci. According to the ninth edition of the fungal dictionary, Shiraia was classified under Dothideales incertae sedis (Kirk et al., 2008). Previous taxonomic classifications of Shiraia were mainly based on morphological characteristics of the ascostromata, ascus, ascospores, etc. However, several traditional morphological features are not class unique and DNA sequence comparisons are important to define the class. Recent studies sequencing the 18S rDNA and ITS-5.8S rDNA regions indicated that Shiraia should be classified under the order Pleosporales (Cheng et al., 2004). More recently, a new family, Shiraiaceae, has been proposed with Shiraia as the type genus. Its epitype was redescribed based on morphological characters and partial LSU-rDNA, EF, and RPB gene sequencing data, suggesting that it is a new family of Pleosporales (Liu et al., 2013). With the availability of more microbial genome sequencing data, researchers have begun to study phylogeny at the genomic level. However, the phylogeny of S. bambusicola has not been analyzed based on data at the genomic level. A phylogenetic analysis based on a small number of concatenated genes in any genome has a high probability of supporting conflicting topologies, while analysis of the entire data set of concatenated genes may provide a single, fully resolved species tree with maximum support (Rokas et al., 2003).
Fungal genomics and comparative analysis of different genomes are helpful in understanding phylogenetic and evolutionary relationships, gene function, lifestyle and strategies, biocontrol mechanisms, pathogenicity, and the development and utilization of secondary metabolites. Given the importance of these studies, we sequenced the S. bambusicola S4201 genome with Illumina HiSeq sequencing technology combined with PacBio single molecular long-read sequencing. A rich repertoire of CAZymes and pathogenesis-related genes were discovered. The taxonomic position of S. bambusicola was reconfirmed based on genomic data. A large set of secondary metabolic biosynthetic gene clusters and core genes were identified. Furthermore, we identified the HA gene cluster and proposed a new putative biosynthetic pathway. The information contained in this study could be useful for understanding the pathogenicity, taxonomic status, and diversity of the secondary metabolites of S. bambusicola S4201.
Materials and Methods
Strain, DNA, and RNA Isolation
Shiraia bambusicola S4201 was isolated in China from the fruiting bodies of S. bambusicola P. Henn, a strain which has shown excellent HA production and which has been used for sequencing (Zhao et al., 2016). The strain S4201 was incubated in potato dextrose broth (PDB, 200 g/L potato extract, 20 g/L glucose, pH 7.0) medium for 120 h at 28°C under agitation (150 r/min). The fermentation liquid was centrifuged at 3000 r/min for 5 min at room temperature and the supernatant was removed. The mycelium was washed with sterile water and frozen in liquid nitrogen. Genomic DNA was isolated using the cetyl trimethyl ammonium bromide (CTAB) method (Rogers and Bendich, 1994). The quality of the total DNA was confirmed by means of a Qubit Fluorometer, NanoDrop spectrophotometer, and by agarose gel electrophoresis before further processing.
Genome Sequencing and Assembly
The S. bambusicola S4201 genome sequences included short reads from the Illumina library and long reads from a PacBio single-molecule library at the Beijing Genomics Institute (BGI) in Wuhan, China. A library with 410 bp inserts was constructed and sequenced using an Illumina Hiseq 4000 platform. To obtain clean data, 20 low-quality bases, 10% Ns, 15 bp overlap between adapter and duplications in the raw Illumina sequencing data were filtered. Total DNA (10 μg) was used to construct a 20 kb DNA library for sequencing with the PacBio platform. The PacBio data were also cleaned by removing adapter sequences, low-quality polymerase reads, and by discarding trimmed reads with lengths less than 1000 bp. The abundance of 15-mers was measured to obtain a preliminary estimate of the genome size, heterozygosity, and repetitive sequences information. Sequences were assembled using various assembly software and according to the following steps: (i) correcting the subreads to obtain the corrected reads (Proovread 2.12, -t 4 –coverage 60 –mode sr); (ii) assembling the corrected reads (Celera Assembler 8.3, doTrim_initialQualityBased = 1, doTrim_finalEvidenceBased = 1, doRemoveSpurReads = 1, doRemoveChimericReads = 1, -d properties -U); (iii) correcting the data using Illumina short reads (GATK v1.6-13, -cluster 2 -window 5 -stand_call_conf 50 -stand_emit_conf 10.0 -dcov 200 MQ0 ≥ 4) (Kim et al., 2014; Badouin et al., 2015; Faino et al., 2015; Sit et al., 2015; Tsuji et al., 2015).
Gene Prediction and Annotation
The gene components were predicted by using Genewise (Birney et al., 2004), SNAP (Johnson et al., 2008), Genemark-ES (Ter-Hovhannisyan et al., 2008), and Augustus (Stanke et al., 2008) software. rRNA was aligned to an rRNA database or predicted by means of RNAmmer (Lagesen et al., 2007) software. tRNA and tRNA secondary structures were predicted with tRNA scan (Lowe and Eddy, 1997) and sRNA was matched with the Rfam (Gardner et al., 2009) database by using Infernal software. RepeatMasker and RepeatProteinMasker software were employed to examine the repeats present in S. bambusicola S4201. BuildXDFDatabase, RepeatModeler, and Repeatmasker were used for de novo detection. The tandem repeat sequences were identified using Tandem Repeat Finder (Benson, 1999) software. The protein-encoding genes were annotated through BLASTp searches in the Cluster of Orthologous Groups of proteins (COG) (Galperin et al., 2015; Makarova et al., 2015), Gene Ontology (GO) (Sherlock, 2009), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2016), Non-Redundant (NR) Protein Database, SwissProt Databases and the best hits were filtered (E-value < 0.00001). CAZymes database (Levasseur et al., 2013) was used to identify proteins involved in carbohydrate metabolism. Pathogenicity and virulence-associated genes were predicted by means of the PHI database (Torto-Alalibo et al., 2009). CYPs were annotated using the Fungal CYP Database (Fischer et al., 2007).
Comparative Genomics and Phylogenetic Tree
Since S. bambusicola S4201 is a single genus and species, we searched for several closely related species (Leptosphaeria maculans, Parastagonospora nodorum, Paraphaeosphaeria sporulosa, Cercospora zeina, Stagonospora sp. SRC1lsM3a, Ascochyta rabiei, Diplodia corticola, Trichoderma citrinoviride, Neurospora crassa) for evolutionary analysis. All of the related species genome sequences were downloaded from the National Center for Biotechnology Information (NCBI) database. To avoid the effects of alternative splicing, we chose the longest transcripts to represent the coding sequences. Gene families were defined by TreeFam methodology and then clustered with Hcluster_sg software (Li et al., 2006; Ruan et al., 2008). The orthologous groups and single-copy orthologs of these fungi were detected using the Perl script modified according to the programing code of Hacquard et al. (2016). Multiple sequence alignments of protein sequences were generated for each gene family using MUSCLE (Edgar, 2004), which converted the CDS alignments into protein alignments. A maximum likelihood phylogenetic tree was created using TreeBest1 with WAG amino acid substitution model. Synteny analysis and core-pan genes analysis were performed on four fungal species (S. bambusicola, P. sporulosa, P. nodorum, and L. maculans).
Analysis of Core Genes and Gene Clusters Involved in Secondary Metabolism
The web-based prediction tool antibiotics and Secondary Metabolite Analysis Shell (antiSMASH) were used to predict secondary metabolite gene clusters and core genes (Blin et al., 2017). Based on previous RNA-Seq data from S. bambusicola S4201 (NCBI accession numbers: SRR2352154 and SRR2153022), we found the FPKM measurements of core genes which were commonly used to present gene expression levels. The PKS domain structures of different species were assigned according to the Conserved Domain Database (CDD) from the NCBI Search database and antiSMASH. The KS domain is the most conserved and is commonly used to infer the phylogenetic relationship between PKS genes. The predicted KS domains of S. bambusicola S4201 and other fungi were aligned by using ClustalW (Larkin et al., 2007) and the neighbor-joining tree was created with MEGA7, with 1000 bootstrap replicates (Kumar et al., 2016).
Expression Vector Construction, Overexpression, and HPLC Profiling
The promoter GPD1, selection marker gene ben, promoter GPD2, and zinc finger transcription factor gene (zftf, as specialized to be involved in HA biosynthesis) were separately amplified from the S4201 genome and from pDHt-Ben using the primers OE-GPD-F1/OE-GPD-R1, OE-Ben-F1/OE-Ben-R1, OE-GPD-F2/OE-GPD-R2, and OE-zftf-F1/OE-zftf-R1. Then, the GPD1-Ben and GPD2-Zftf fragments were generated by overlapping PCR using PrimeSTAR® Max DNA Polymerase (TaKaRa, Dalian, China) and primers OE-GPD-F1/OE-Ben-R1 and OE-GPD-F2/OE-zftf-R1. In addition, the GPD1-Ben fragment was cloned into the pEASY-Blunt Zero vector (TransGen Biotech) to generate the plasmid pGPD1-Ben. The GPD2-Zftf fragment was subcloned into the PmeI site of pGPD1-Ben, forming the plasmid pOE-zftf. S. bambusicola S4201 was grown in PDA at 28°C for 120 h. The spores were collected in sterile water, and the protoplasts were prepared by adding an enzyme mixture. The plasmids were introduced into S. bambusicola S4201 by the polyethylene glycol-calcium chloride (PEG-CaCl2) transformation method, according to published procedures (Jiang et al., 2014). After culturing for 120 h at 28°C, the transformants were identified by diagnostic PCR and the sequences were analyzed using the DNA of the mycelium. Primer sequences are shown in the supporting information section (Supplementary Table S1). Cultures of 1 × 105 conidia per mL of S4201 and the overexpressing transformants were grown in PDB at 28°C under constant agitation at 150 r/min for 120 h. Next, the mycelia were used for detection of expression by qRT-PCR. The culture supernatant was extracted with ethyl acetate until it turned colorless. Then, the extracts were concentrated to dryness and the residue was dissolved in acetonitrile. The HA and elsinochrome A (EA) concentrations in the extracts were measured by high-performance liquid chromatography (HPLC) using standard reagents. HPLC was performed using an Agilent Technologies 1220 Infinity LC instrument. The HA and EA of S. bambusicola S4201 and the overexpression transformant were analyzed by HPLC at 30°C using a C18 column (5 μm, 4.6 × 250 mm) (SunFireTM, Waters, United States), with a flow rate of 1 mL/min and injection volume of 10 μL.
Quantitative Real-Time PCR Analysis
The glyceraldehyde-3-phosphate dehydrogenase gene (GAPDH) was used as the internal reference. Total RNA was isolated using TRIzol reagent according to the instructions of the manufacturer (Vazyme, Nanjing, China). The RNA quality was assessed with 1.0% agarose gels and verified using NanoDrop 2000 (Thermo Fisher Scientific, United States). cDNA was synthesized using the HiScript II First Strand cDNA Synthesis Kit (+ gDNA wiper) (Vazyme, Nanjing, China). qRT-PCR was performed using a StepOnePlusTM Real-Time PCR System (Applied Biosystems, United States) and AceQ qPCR SYBR Green Master Mix (Vazyme, Nanjing, China). The results were calculated using the 2–ΔΔCt method. All the gene-specific primers are listed in Supplementary Table S2. All the experimental data were obtained from three biological replicates.
Results
De novo Genome Sequencing, Assembly, and Annotation
We isolated endophytic S. bambusicola S4201 fungi from the fruiting bodies of S. bambusicola P. Henn and demonstrated that it produced HA, based on HPLC analysis (Zhao et al., 2016). To gain insight into the pathogenicity, classification, and secondary metabolites of S. bambusicola S4201, we de novo sequenced and assembled its genome. By integrating Illumina HiSeq and PacBio single molecule long-read sequencing, we generated 2649 Mb of raw data and 4359 Mb of polymerase read bases. After removing the adapter sequence and low-quality reads, we obtained 2053 Mb of clean data and 4335 Mb of subread bases. The final S. bambusicola S4201 genome assembly included 32 Mb in 57 contigs, with a contig N50 of 1,565,644 bp (Figure 2). The genomic information was compared with that of Shiraia sp. slf14 (GenBank: AXZN00000000.1, no annotation, unpublished results) in the NCBI database (Table 1). Moreover, the gene length distributions and open reading frames of the genome were predicted based on the assembly sequences (Supplementary Figure S1 and Supplementary Table S3). Non-coding RNA (ncRNA) statistics are shown in Supplementary Table S4. The repeats, long terminal repeats (LTRs), long interspersed elements (LINE), and short interspersed elements (SINE) are shown in Supplementary Tables S5, S6. The genome sequence of S. bambusicola S4201 has been deposited at GenBank under accession number SRR8379567.
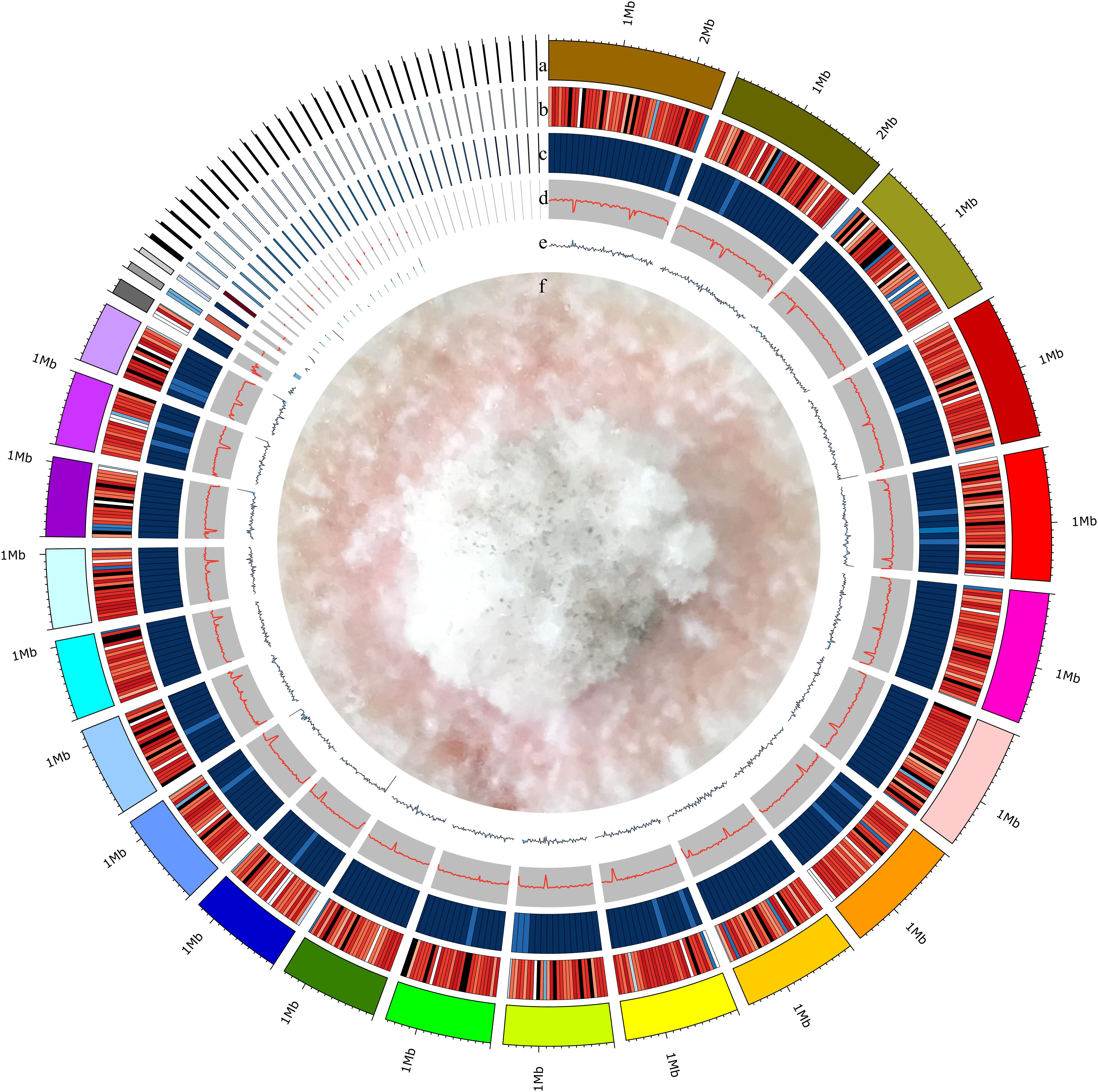
Figure 2. Circular representation of the Shiraia bambusicola genome. The following data are shown (from outer to inner): (a) Genome; sorted by length. (b) Gene density; gene number in 50,000 bp non-overlapping windows. (c) ncRNA density; ncRNA number in 100,000 bp non-overlapping windows. (d) GC; GC rate in 20,000 bp non-overlapping windows. (e) GC skew; GC skew in 20,000 bp non-overlapping windows. (f) The mycelia of Shiraia bambusicola S4201.
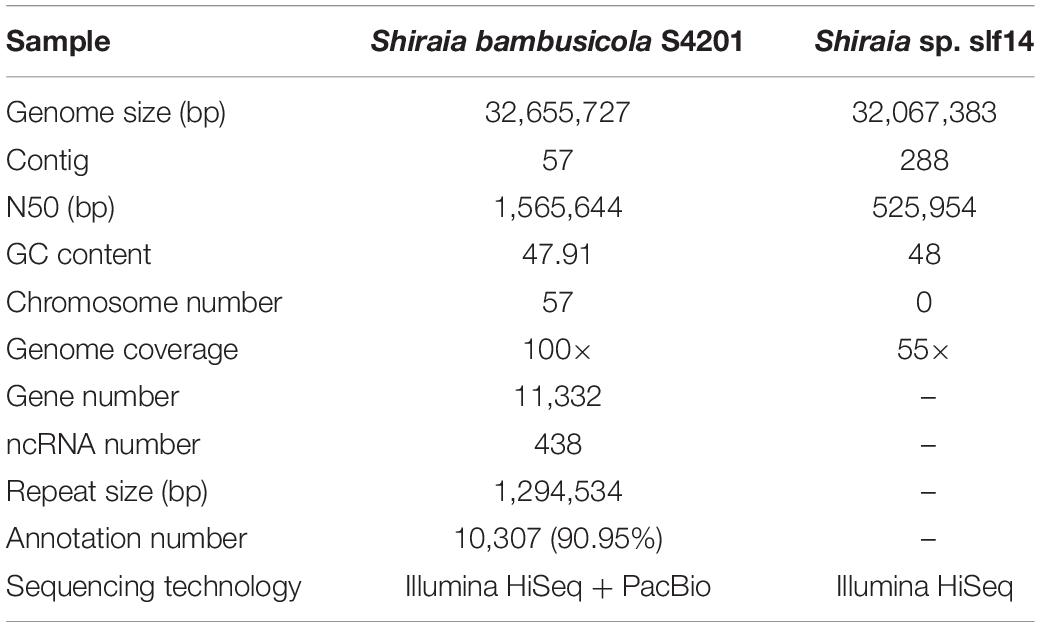
Table 1. Characteristics of the genome of Shiraia bambusicola.
To conduct functional annotation of the S4201 gene model, we used the blast search function to enter the putative protein-coding sequences into the GO, KEGG, COG, Swiss-Prot, Trembl, GenBank NR, EggNOG (Huerta-Cepas et al., 2016), and TransportDB databases. There were 6358 (56.10%) annotated genes obtained for the three main GO categories of biological process, cellular component, and molecular function, including 48 sub-categories (Supplementary Figure S2). In total, 4368 (38.54%) genes were annotated and assigned to 45 different KEGG pathways (Supplementary Figure S3). “Carbohydrate metabolism” was the most enriched pathway, followed by “Amino acid metabolism” and “Translation.” In summary, 1318 (11.63%) genes were annotated into the COG database (Supplementary Figure S4). Among the 25 COG functional categories, the cluster for “General function prediction only” was the largest, followed by “Amino acid transport and metabolism” and “Carbohydrate transport and metabolism.” Only a few genes were assigned to the “Extracellular structures,” “Chromatin structure and dynamics,” and “RNA processing and modification” groups.
Genes Involved in Pathogenicity
To produce a successful infection, phytopathogenic fungi often have to break down plant cell walls and penetrate into host tissues by means of CAZymes (Yong et al., 2014). The S. bambusicola S4201 genome encodes 414 putative CAZymes, including 166 glycoside hydrolases (GHs), 52 glycosyltransferases (GTs), nine polysaccharide lyases (PLs), 13 carbohydrate esterases (CEs), 72 auxiliary activities (AAs), and 102 carbohydrate-binding modules (CBMs). A heatmap based on the classification of CAZymes of 20 fungal species (N. crassa OR74A, Aspergillus oryzae RIB40, Debaryomyces hansenii CBS767, Fusarium fujikuroi IMI 58289, Hypocrella siamensis MTCC 10142, L. maculans JN3, Melanopsichium pennsylvanicum 4, Thielavia terrestris NRRL 8126, Thermothelomyces thermophila ATCC 42464, Ustilago bromivora UB2112, Xanthophyllomyces dendrorhous, Yarrowia lipolytica CLIB122, Zygosaccharomyces bailii CLIB 213, Zymoseptoria tritici ST99CH_1A5, Z. tritici ST99CH_1E4, Arthrobotrys oligospora ATCC 24927, Aspergillus nidulans FGSC A4, Aspergillus niger CBS 513.88, Blumeria graminis f. sp. tritici, S. bambusicola S4201) is shown in Figure 3. The AA coding ability of S. bambusicola S4201 was higher than that of other strains, and the CBM coding ability was second only to A. oligospora ATCC 24927. However, its GT coding ability was the weakest, lower than other selected strains. The proteins containing CAZy domains in the GH, PL, and CE groups may act as plant polysaccharide degradation (PDD) enzymes (Han et al., 2016). Regarding the PDD enzyme families, 99 sequences were predicted, distributed in 34 families (Supplementary Table S7). As a component of the plant cell wall and the intercellular spaces, pectin can provide nutrients to pathogenic fungi. S. bambusicola S4201 has many candidate pectinases and includes almost all pectinase families known in fungi, including PL1, PL3, PL4, GH78, GH88, GH95, GH105, and GH115. Moreover, some putative pathogenic factors were predicted, such as cutinase genes, which may be needed to catalyze plant cuticle degradation. In addition, proteins belonging to the GH6, GH7, GH39, and GH51 families, involved in the degradation of cellulose, hemi-cellulose, and pectin in plant cell walls, were also predicted. Eight hundred ninety genes in S. bambusicola S4201 were putatively involved in pathogenicity and virulence when analyzed with the PHI database, and this accounts for 7.85% of the total predicted genes (Supplementary Table S8). One thousand twenty-six putative P450s (9.05%) were predicted in S. bambusicola S4201 (Supplementary Table S9). In total, 74 major facilitator superfamily (MFS) transporters (Supplementary Table S10) and 46 ATP-Binding Cassette (ABC) transporters (Supplementary Table S11) were predicted.
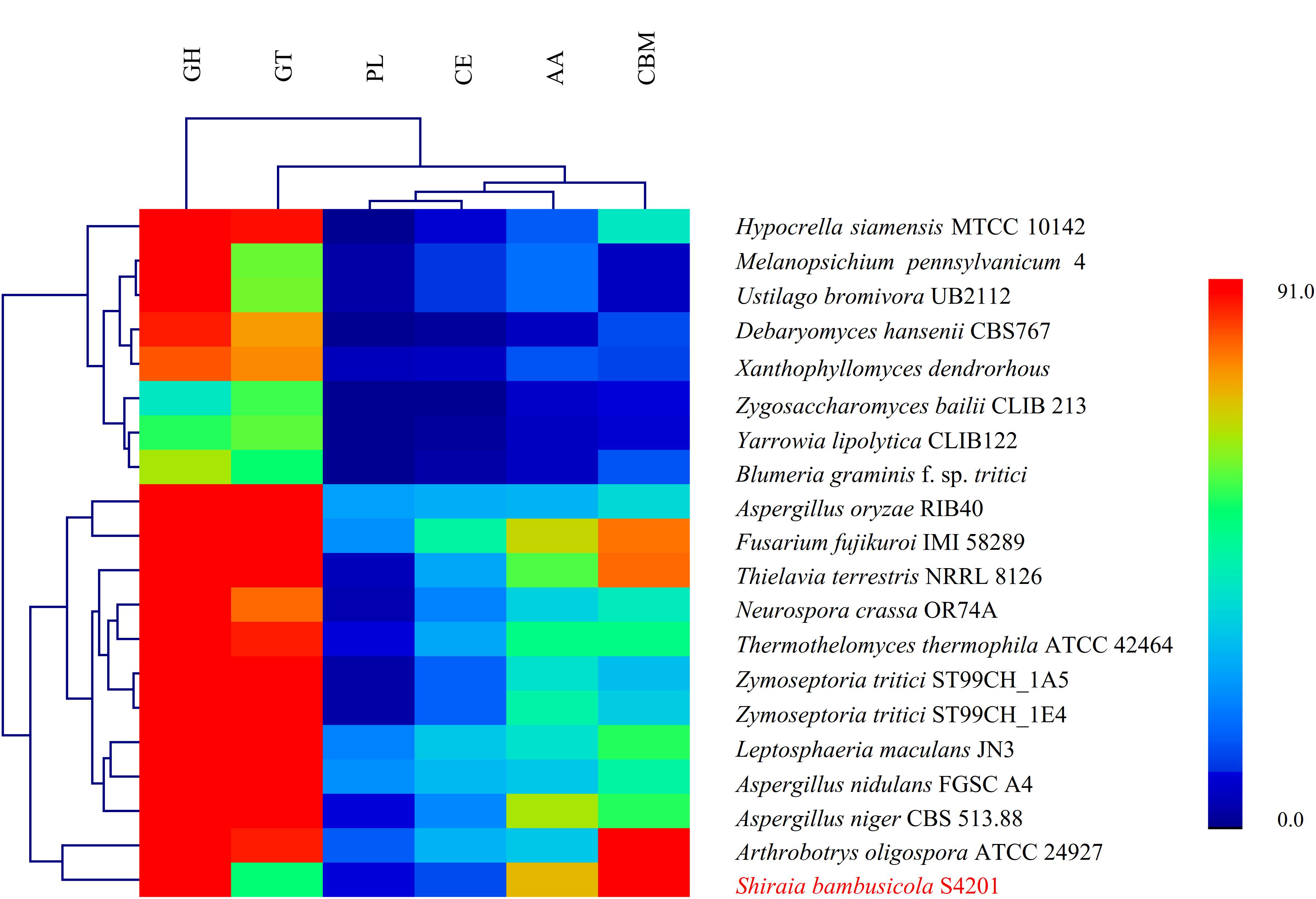
Figure 3. Hierarchical clustering of CAZyme in the genomes of Shiraia bambusicola and 19 other fungal species. The enzyme families are classified as follows: GH, glycoside hydrolases; GT, glycosyl transferases; PL, polysaccharide lyases; CE, carbohydrate esterases; AA, auxiliary activities; and CBM, carbohydrate-binding modules. The family numbers are based on the carbohydrate-active enzyme database. A color scale indicates the numbers of the different enzymes in each genome, from lowest (blue) to highest (red).
Comparative Genomics and Classification
In S. bambusicola S4201 and nine other selected fungal species (L. maculans, P. nodorum, P. sporulosa, C. zeina, Stagonospora sp. SRC1lsM3a, A. rabiei, D. corticola, T. citrinoviride, N. crassa), a total of 120,799 genes clustered into 10,851 gene families, including 3423 (31.55%) gene families shared by all species and 2105 (19.40%) that were single copy orthologs (Supplementary Table S12). The differences and quantities of ortholog groups among the fungi are shown in Supplementary Figure S5. The taxonomic status of S. bambusicola among other nine fungal species was evaluated. Based on highly conserved single copy orthologs, the phylogenetic tree (Figure 4) was constructed with the maximum likelihood method. The S. bambusicola S4201 genome was further characterized by comparative gene functional analyses with the following closely related species: P. sporulosa, P. nodorum, and L. maculans. Comparative analyses of the four available genomes revealed that P. nodorum had the largest number of genes. Synteny analysis showed that S. bambusicola S4201 and P. nodorum genome shared many large areas of synteny (Supplementary Figure S6) and they also share more homologous proteins. Comparative analysis of core and pan genes among the four species showed that there are 3480 genes classified as core genes, 35,395 genes classified as pan genes, and 5275 genes classified as dispensable genes (Supplementary Figure S7). The heatmap of dispensable genes in these species suggests that S. bambusicola S4201 has the fewest species specific genes (Supplementary Figure S8). It shows that S. bambusicola S4201 is more closely related to P. nodorum (Phaeosphaeriaceae, Pleosporales, Dothideomycetes) than to other species. The analysis revealed that S. bambusicola S4201 belongs among the species of Pleosporales, Dothideomycetes.
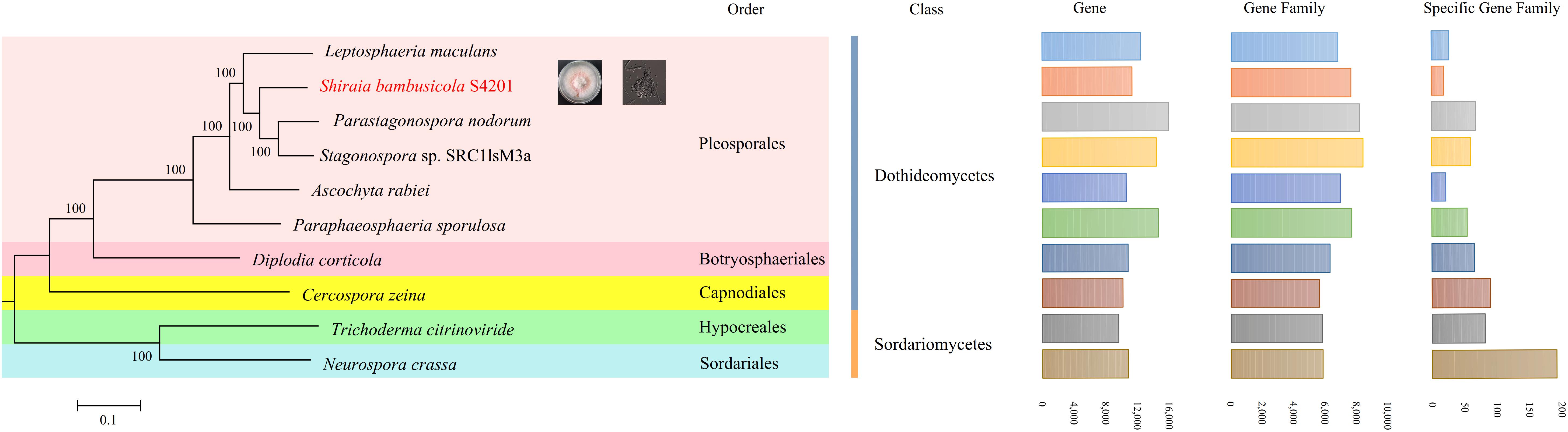
Figure 4. A maximum likelihood phylogenetic tree was inferred based on the highly conserved single copy orthologs of ten fungal genomes. The numbers on the branch points of the phylogenetic tree represent the bootstrap values (1000 replicates). Scale lengths represent the genetic distance. The 10 fungi are marked with different colors. The statistics for gene numbers, family numbers, and unique families in the 10 fungal species are shown.
Secondary Metabolites
Filamentous fungi can produce many secondary metabolites, such as bioactive compounds or mycotoxins, which have been used for the synthesis of pharmaceuticals. In fungi, the core genes, accessory genes, regulators, transporters, and other genes involved in the biosynthesis and modification of secondary metabolites are organized in clusters (Daub and Chung, 2009). Using the scaffolds as the query sequences for the antiSMASH 4.1.0 platform, 73 putative secondary biosynthetic gene clusters were predicted. In total, 15 polyketide synthases (PKS, 14 T1PKS, and one T3PKS), six non-ribosomal peptide synthases (NRPS), one linaridin, two terpenes, one indole, one cf_fatty_acid, 28 cf_putative, and two other gene clusters were identified in the S. bambusicola S4201 genome, distributed among 20 scaffolds. Cluster 22 shared 26% gene similarity with the patulin biosynthetic gene cluster, cluster 60 is likely to be an asperfuranone biosynthetic gene cluster (27% of genes show similarity) and cluster 72 acts as a cyclochlorotine biosynthetic gene cluster (25% of genes show similarity). These results indicate that these predicted gene clusters may produce the mentioned secondary metabolites in S. bambusicola S4201.
To further understand the expression patterns of core biosynthetic genes involved in secondary metabolite biosynthesis, we estimated the expression of these genes in fragments per kb per million reads (FPKM) using previous RNA-seq data from S. bambusicola S4201 cultured for 82 h in PDB medium with Triton X-100 (Zhao et al., 2016). The secondary metabolite biosynthetic gene clusters are shown in Table 2. Phylogenetic analysis based on the KS domain amino acid sequences of PKSs in S. bambusicola S4201 and the products of known PKSs, was divided according to three main classes of PKSs, including highly reduced (HR) PKSs, partially reduced (PR) PKSs, and non-reduced (NR) PKSs (Figure 5). The reduced PKSs contain the reductive domains dehydratase (DH), enoyl reductase (ER), and keto-reductase (KR), while the NR-PKSs do not. ctg2_orf583, ctg16_orf306, ctg9_orf6, ctg18_orf232, ctg13_orf57, ctg17_orf70, and ctg12_orf33 were grouped with lovastatin and compactin synthase from Aspergillus terreus and Penicillium citrinum in the HR-PKS class. On the other hand, ctg21_orf150, ctg7_orf10, and ctg2_orf331 were nested in the PR-PKS clade. In addition, ctg1_orf376, ctg20_orf61, and ctg2_orf265 were distributed in the NR-PKS class, which include the polyketides cercosporin, aflatoxin, elsinochrome, and a putative polyketide involved in HA biosynthesis (ctg14_orf277).
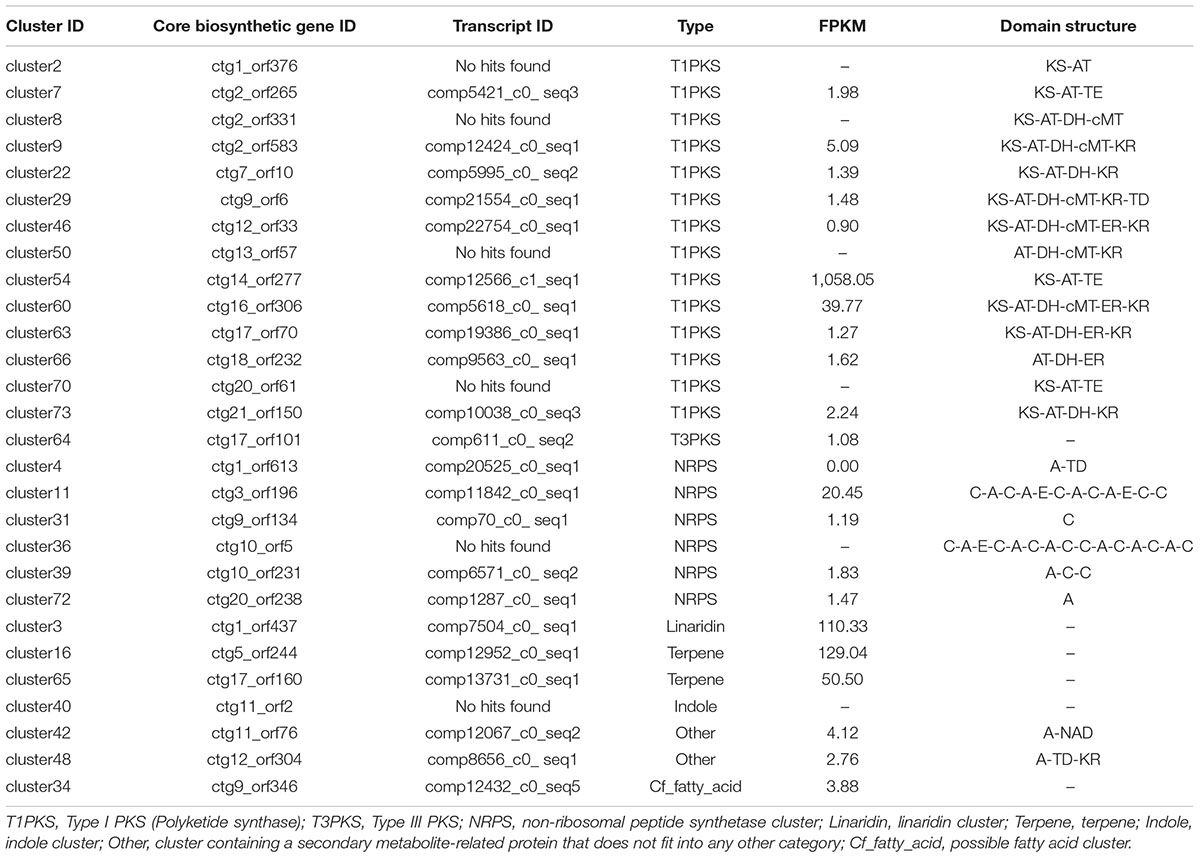
Table 2. Secondary metabolite biosynthetic gene clusters in Shiraia bambusicola S4201.

Figure 5. Phylogenetic analysis of the KS domain amino acid sequences of PKSs in Shiraia bambusicola S4201 and other fungal species. The numbers on the branch points of the phylogenetic tree represent the bootstrap values (1000 replicates). Bootstrap support values > 50% are shown above each branch. Scale lengths represent the genetic distance. Toxins or other polyketides are shown together with the fungal species. The PKS domain structures of different species are shown. The classification of their PKSs is also shown in the rightmost part of the figure.
The putative HA biosynthesis genes were classified into cluster 54, which was located on scaffold 14 (location: 1101201–1146665 nt) and included 13 genes (Figure 6). The genes in cluster 54 probably encode a FAD/FMN-dependent oxidoreductase (ctg14_orf271), a hydroxylase (ctg14_orf272), a zinc finger transcription factor (ctg14_orf273), an o-methyltransferase (ctg14_orf274), an MFS transporter (ctg14_orf275), an o-methyltransferase/FAD-dependent monooxygenase (ctg14_orf276), a polyketide synthase (ctg14_orf277), a dynamin GTPase domain (ctg14_orf278), and hypothetical proteins (ctg14_orf279–ctg14_orf283) (Supplementary Table S13).

Figure 6. Putative hypocrellin A biosynthetic gene cluster in Shiraia bambusicola S4201 and Shiraia sp. slf14. (A) Putative hypocrellin A biosynthetic gene cluster in Shiraia bambusicola S4201. Cluster 54, with 13 genes, is located on scaffold 14 (1101201–1146665 nt) and was identified by using antiSMASH. Coding regions are indicated with arrows and are putatively involved in HA biosynthesis or have unknown functions. (B) Putative hypocrellin A biosynthetic gene cluster in Shiraia sp. slf14 based on the National Center for Biotechnology Information database (GenBank: KM434884.1).
Overexpression of the Zinc Finger Transcription Factor zftf
A powerful approach to enhance the production of secondary metabolites is to overexpress Zn(II)Cys6-type pathway-specific transcription factors embedded in the corresponding gene clusters (Bennett, 2005; Bergmann et al., 2007). To determine the function of zftf, we cloned it into a plasmid, then transformed S. bambusicola S4201 with pOE-zftf, and the OE mutants were selected by diagnostic PCR amplification of the expression cassette. HPLC analysis with standard reagents (Supplementary Figure S9) showed that HA production by the OE mutant (206.75 mg/L) was higher than in wild type (82.60 mg/L). The expression levels of zftf in the mycelia were analyzed by quantitative real-time PCR (qRT-PCR), and the OE mutant showed more than threefold upregulation on average. These results show that the zinc finger transcription factor zftf plays an important regulatory role in HA production.
Quantitative Real-Time PCR Analysis of Several Genes
Seven genes (encoding polyketide synthase, o-methyltransferase/FAD-dependent monooxygenase, o-methyltransferase, FAD/FMN-dependent oxidoreductase, hydroxylase, MFS transporter, and zinc finger transcription factor) involved in HA biosynthesis were selected for qRT-PCR analysis to validate changes in gene expression. qRT-PCR analysis of total RNA extracted from wild type and the OE mutant grown in PDA showed that expression of these genes was upregulated in the OE mutant (Figure 7) indicating that the ZFTF transcriptional activator controls HA production by controlling gene transcript levels. Based on the previous RNA-Seq data (Zhao et al., 2016), the heatmap analysis of the expression of these genes in S4201-D1 and S4201-W was consistent with that in S4201-W and OE mutant. S. bambusicola S4201-W shows abundant HA production, while the S4201-D1 mutant does not. Thus, the HA biosynthesis core gene cluster is apparently coregulated through zftf, which encodes a Zn(II)Cys6 transcriptional activator, as expression of relevant genes was enhanced in the OE mutant.
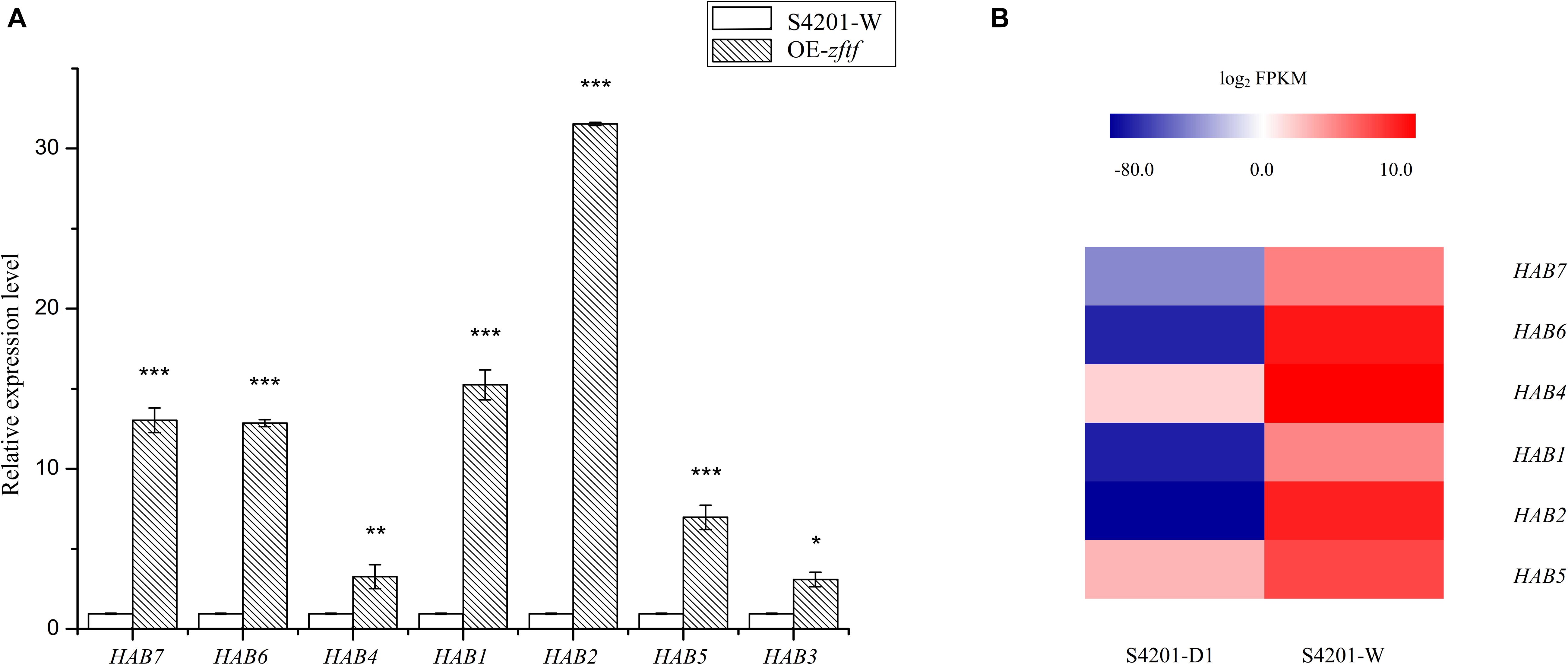
Figure 7. Expression of genes putatively involved in HA biosynthesis. (A) Quantitative RT-PCR validation. The relative mRNA expression levels were normalized with respect to the internal reference gene GAPDH and expression by S4201-W (control) was given an arbitrary value of 1. The abscissa shows the genes that encode polyketide synthase, o-methyltransferase/FAD-dependent monooxygenase, o-methyltransferase, FAD/FMN-dependent oxidoreductase, hydroxylase, major facilitator superfamily (MFS) transporter, and zinc finger transcription factor. *P < 0.05, **P < 0.01, ***P < 0.001 vs control group. (B) Heatmap of differentially expressed genes based on previous RNA-Seq data (Zhao et al., 2016). Shiraia bambusicola S4201-W shows abundant HA production, while the S4201-D1 mutant does not. A color scale indicates the expression levels of the different genes, from lowest (blue) to highest (red).
Discussion
Shiraia bambusicola is an important bamboo ascomycete pathogen. In this study, we sequenced, assembled, and analyzed the genome of this pathogen. The genome data are the initial step to understand the biology of S. bambusicola.
The chemical composition of bamboo fibers is mainly lignin, cellulose, and hemicellulose. Lignin accounts for the hardness and yellowness of bamboo fibers and is present at different concentrations in different layers of the cell wall (Khalil et al., 2012). In general, fungal pathogens possess pathogenic pathways which mediate cell wall degradation to enter host cells, interact with host cells, secrete toxins, and transport. During the infection stage, the polysaccharide content of the host cell wall is destroyed mainly through the action of CAZymes (Ospina-Giraldo et al., 2010), and these enzymes can also be used to utilize nutrients from the host cell. Analysis of CAZymes provides useful information about the lifestyles of fungi (Ohm et al., 2012). AAs include redox enzymes that act in conjunction with CAZymes. The AA group presently includes nine families of ligninolytic enzymes and six families of lytic polysaccharide monooxygenases (Levasseur et al., 2013). Among the 20 species analyzed, the AA content in S. bambusicola S4201 was the highest. The AAs aid in the hydrolysis of lignin, which are the main component of bamboo, providing a basis for S. bambusicola’s parasitism of bamboo. Apart from the catalytic modules, about 7% of CAZymes include CBMs, the most common non-catalytic modules linked to cell wall hydrolytic enzymes (Cantarel et al., 2009). They play an important role in the enzymatic hydrolysis of plant structures, in storage and in the degradation of other insoluble polysaccharides (Boraston et al., 2004). The higher content of CBMs in S. bambusicola S4201 may help it hydrolyze insoluble polysaccharides such as cellulase in bamboo, damaging cell walls and facilitating parasitism of this plant. Organisms with very large genomes that can synthesize complex cell walls or use glycosylation of small molecules to regulate biological activity have many GTs. By contrast, organisms that have experienced significant gene loss over the course of evolution and which have become obligate symbionts or obligate parasites appear to have little or no detectable GT genes (Lairson et al., 2008). Thus, it is of interest that the number of GTs in S. bambusicola S4201 was significantly less than in the other 19 species. As mentioned above, “Carbohydrate metabolism” was the most enriched pathway in KEGG pathways and lots of genes assigned to the “Carbohydrate transport and metabolism” in COG categories. Therefore, a large number of genes involved in carbohydrate transport and metabolism of S. bambusicola S4201 may be closely related to its pathogenicity. In the future, the pathogenicity analysis can be expanded as follows: first, by selecting at least four RNA samples from different infection stages or infection sites for transcriptome sequencing analysis. And second, by knocking out the selected pathogenicity-related genes to determine their relationship with S. bambusicola pathogenicity.
Dothideomycetes is regarded as the largest and most diverse class of Ascomycete fungi, and comprises 11 orders, 90 families, 1300 genera, and more than 19,000 known species. The species are taxonomically classified into three subclasses, 11 orders: Capnodiales, Dothideales and Myriangiales (Dothideomycetidae); Hysteriales, Jahnulales, Mytilinidiales and Pleosporales (Pleosporomycetidae); Botryosphaeriales, Microthyriales, Patellariales, and Trypetheliales (Incertae sedis). Among them, the most diverse fungal order in Dothideomycetes is Pleosporales, which represent roughly a quarter of all Dothideomycetous species (Kirk et al., 2008). In order to determine the classification of S. bambusicola, nine fungal species were used for phylogenetic construction using the complete genome sequencing data downloaded from the NCBI database. The results from the phylogenetic analysis were basically consistent with previous studies (Liu et al., 2013; Shen et al., 2015), which showed that S. bambusicola is located in the order Pleosporales. The inferred phylogeny indicated that Stagonospora sp. SRC1lsM3a and P. nodorum were most closely related to S. bambusicola; thus, they may share a common pathogenic fungal ancestor. Further studies are needed to clarify the evolutionary relationship between S. bambusicola and the proximal species. This is the first study supporting the taxonomic status of S. bambusicola at the genomic level. The analysis of synteny between four fungi in Pleosporales indicated that the S. bambusicola S4201 and P. nodorum genomes shared a high degree of synteny, followed by P. sporulosa and L. maculans. Although there is a degree of synteny among related species, we observed significant chromosome rearrangement. At the same time, S. bambusicola S4201 had the fewest species specific genes, suggesting that it was relatively conservative during its evolution.
In the secretion and transport stages, secondary metabolites act as virulence factors, and transport is also important for pathogenesis. These secondary metabolites, particularly mycotoxins in S. bambusicola, may be responsible for its adaptation to an exclusively parasitic life cycle. There are mainly two transport systems, ABC-transporter (Gao et al., 2019) and MFS-transporter (Wang et al., 2018). These analyses provide some insights into the role of CAZymes and secondary metabolites in the parasitic lifestyle of S. bambusicola S4201. Seventy-three secondary metabolite biosynthetic gene clusters were predicted in S. bambusicola S4201, 15 of which were polyketide synthase gene clusters. Numerous secondary metabolites have already been found (Liu et al., 2018), indicating there is an enormous potential for production of secondary metabolites by S. bambusicola. Based on the FPKM values in the previously reported RNA-Seq, we estimated the possible expression of the core genes in various secondary metabolite biosynthetic gene clusters, and the genes were generally believed to be expressed when the FPKM value ≥ 0.5. Nevertheless, fungal polyketides are complex to synthesize, due to the length of the polyketide chains, the degree of reduction of intermediates, cyclization and release of products, etc. Therefore, it is difficult to predict the final synthetic metabolic products based only on the DNA or amino acid sequences. First, uncertainty regarding repeated catalysis by polyketide synthase and unknown synthesis processes limit the ability to make predictions based on gene sequences alone. Second, many secondary metabolites are produced in very low amounts and are difficult to detect in wild fungi or are unexpressed in laboratory conditions, so the synthesis genes and products cannot be connected.
To study the phylogenetic relationship of PKSs in the genome of S. bambusicola, the structures of the synthetic products and the PKS reduction types, a phylogenetic tree was constructed based on the ketoacyl synthase domain (KS domain) amino acid sequence of the polyketide synthases of S. bambusicola and 16 other polyketide synthases. Because some of the predicted polyketide synthases of S. bambusicola were similar to the polyketide synthases producing known compounds, and clustered in the phylogenetic tree of the same branch, we speculated that S. bambusicola can produce the same compounds or similar structural analogs (Figure 5). We selected one of these compounds, EA, for HPLC detection. EA was detected when S. bambusicola S4201 was cultured in PDB for 120 h (Supplementary Figure S9). Since S. bambusicola can produce EA and HA, and their structures are similar (Figure 1), we believe that EA is also a type of photosensitizer and virulence factor that can cause bamboo disease.
Some HA biosynthetic genes in the cluster we predicted by antiSMASH differed from the ones in Shiraia sp. slf14 (GenBank: KM434884.1). Compared with Shiraia sp. slf14, the gene clusters we predicted lacked the NADPH-dependent oxidoreductase gene, MFS gene, fasciclin gene, putative multicopper oxidase gene, and three hypothetical proteins genes. In contrast, they included a dynamin GTPase domain, an unknown gene, and four hypothetical proteins genes. Comparison of putative HA biosynthetic gene cluster in S. bambusicola S4201 and Shiraia sp. slf14 is shown in Figure 6. In order to finally determine the function of these genes and whether they are involved in HA biosynthesis, further experiments such as gene knockout are still needed. The putative S. bambusicola S4201 PKS (cluster 54_ctg14_orf277) involved in HA biosynthesis are clustered in the same branch as the Cercospora nicotianae PKS, and their molecular structure and biosynthetic gene clusters are similar. Comparing the CTB and HA cluster genes, NADPH-dependent oxidoreductase (CTB6) and FAD-dependent monooxygenase (CTB7) appeared to be unique to the CTB cluster. Considering the function of these enzymes, it makes sense that they are absent in the HA cluster, given that these structural features are non-existent (Newman and Townsend, 2016). Future studies are needed to determine whether these genes participate in HA biosynthesis.
The Zn(II)Cys6 family of transcription factors can regulate various cellular processes in fungi (Thieme et al., 2018). When the zftf gene was overexpressed, expression of the genes in the putative HA cluster was increased, suggesting that the zinc finger transcription factor zftf plays a major role in the regulation of this pathway. The HPLC results also proved that overexpression of zftf increases HA production. A hypothetical HA biosynthetic pathway has been proposed based on the latest studies on cercosporin (Newman and Townsend, 2016) and elsinochrome C (Chooi et al., 2017), but it still needs to be validated with additional experiments. Figure 8 is a hypothetical flow diagram showing the biosynthesis of HA from substrates to its release by cells, in which the pathways are annotated based on the KEGG database. Due to the wide source of substrates, not all pathways are shown. Acetyl-CoA and malonyl-CoA are the precursors of polyketide biosynthesis, including HA. First, polyketide synthase synthesizes the common intermediate nor-toralactone. Second, o-methyltransferase/FAD-dependent monooxygenase methylates the intermediate at the OH-2 position and opens the pyrone ring, thereby introducing a C6-OH. O-methyltransferase/FAD-dependent monooxygenase is an unusual didomain enzyme with an o-methyltransferase domain and a flavin-dependent monooxygenase domain, and it has unique coupled activities. Third, o-methyltransferase methylates the nascent C6-OH. Fourth, FAD/FMN-dependent oxidoreductase catalyzes the dimerization, producing the perylenequinone carbon core. However, the downstream components and modification reactions in this pathway are still elusive. The HA in the fungal cell may be reduced to a non-toxic form that can minimize cell damage by oxidizing agents. Once synthesized, HA must be exported out of the cell by an MFS transporter. Once it is released, HA may spontaneously re-oxidize to its photoactive form. The identification of the HA pathway in S. bambusicola provides an opportunity to further study the biosynthesis of perylenequinones.
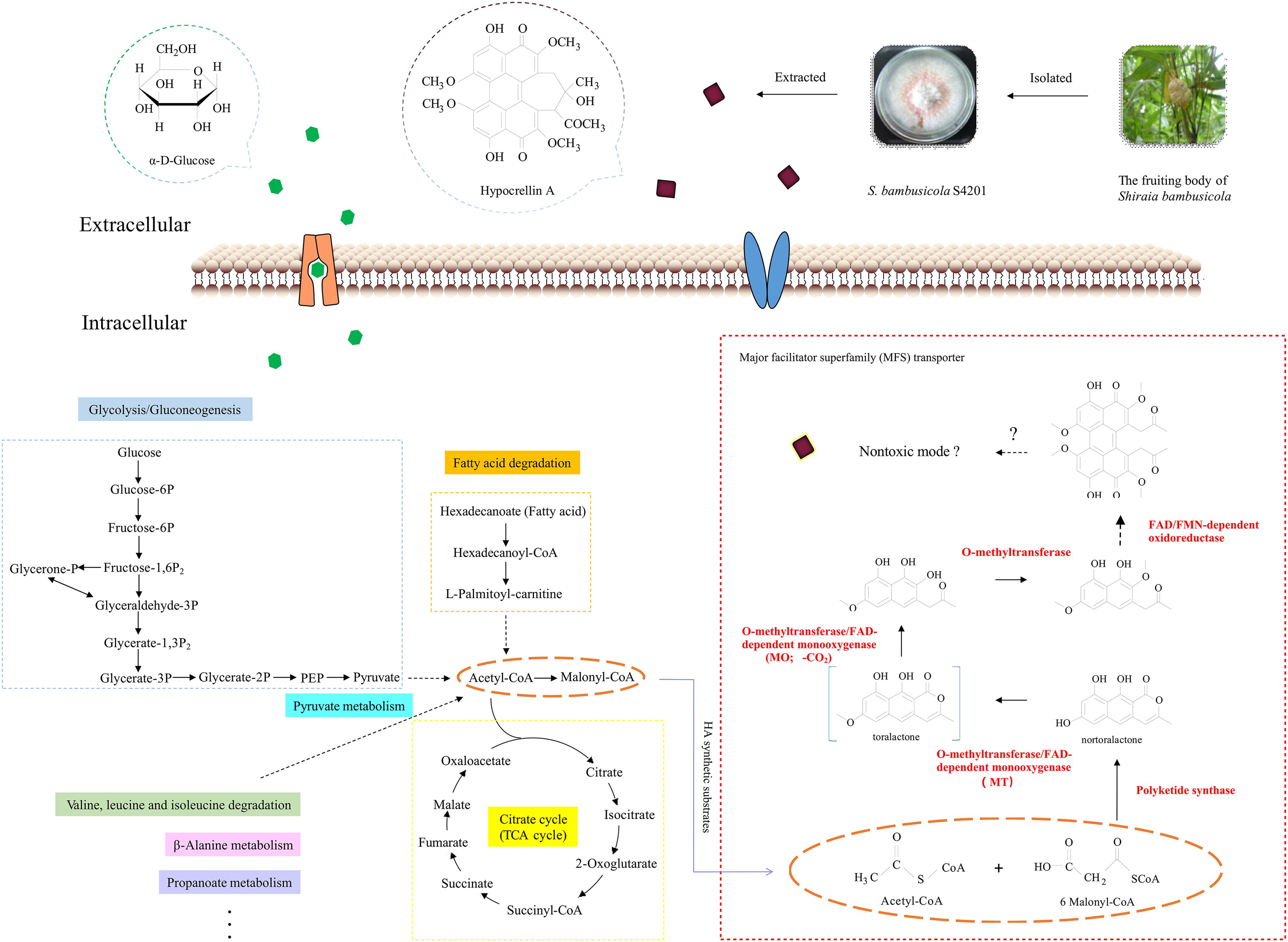
Figure 8. Schematic diagram of the central carbons of hypocrellin A showing its synthesis in Shiraia bambusicola. This flow chart simulates the process of HA synthesis. The highlighted and dotted lines on the left indicate the sources of HA substrates, and the genes marked with red on the right were upregulated in the transcriptome analysis we performed (Zhao et al., 2016). The proposed functions of the proteins encoded by the HA biosynthesis gene cluster are based on the biosynthesis studies of cercosporin (Newman and Townsend, 2016) and elsinochrome C (Chooi et al., 2017). The orange dotted lines represent the substrates for HA biosynthesis. The broken arrows represent putative steps, in which the enzymes involved are still unclear.
Current studies are mainly focused on the fermentation of S. bambusicola and the extraction of secondary metabolites. Future efforts should focus on identifying the genes involved in HA biosynthesis, the intermediate metabolites, and the synthesis pathways by gene knockout experiments and expression of heterologous proteins.
Conclusion
Shiraia bambusicola is an important parasitic pathogen that grows on the twigs of bamboos and which has traditionally been considered to have anti-inflammatory and antiviral properties. We have de novo sequenced and assembled the genome of S. bambusicola S4201, which may serve as a basis for understanding its pathogenicity and exclusive parasitic lifestyle. S. bambusicola is capable of encoding a large and diverse set of CAZy, primary and secondary metabolism enzymes and transporters. The genome comparative study facilitated our understanding of the phylogeny of S. bambusicola and the evolutionary relationships between S. bambusicola and other sequenced species. Numerous secondary metabolite biosynthetic gene clusters were found. In addition, biosynthetic gene clusters for unknown products show the enormous potential that exists for drug discovery in S. bambusicola. Overexpression of the transcription factor zftf increased the production of HA by regulating the expression levels of other genes in the HA biosynthetic gene cluster. A new putative HA biosynthetic pathway was proposed. Our analyses provide some new insights into the pathogenicity, phylogeny, and secondary metabolites of S. bambusicola. In the future, this study will be an important resource for gene discovery and to increase HA production through further genetic manipulation.
Data Availability Statement
The genome data datasets generated for this study can be found in the NCBI Repository with accession number SRR8379567. The RNA-Seq datasets from S. bambusicola S4201 generated for this study can be found in the NCBI Repository with accession numbers SRR2352154 and SRR2153022.
Author Contributions
S-LC, S-ZY, and NZ conceived and designed the experiments. NZ, DL, and B-JG performed the experiments. NZ and XT analyzed the data. XL contributed reagents, materials, and analysis tools. NZ wrote the manuscript.
Funding
This study was supported by the funds of the National Key Technology Research and Development Program of the Ministry of Science and Technology of China (No. 2012BAD36B05) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are sincerely grateful to the referees whose comments made our results more accessible to the readers. We are thankful to Dr. Cheng-Shu Wang (Institute of Plant Physiology and Ecology, SIBS, CAS) for donating plasmid pDHt-Ben.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2020.00643/full#supplementary-material
Footnotes
References
Ali, S. M., and Olivo, M. (2002). Efficacy of hypocrellin pharmacokinetics in phototherapy. Int. J. Oncol. 21, 1229–1237.
Badouin, H., Hood, M. E., Gouzy, J., Aguileta, G., Siguenza, S., Perlin, M. H., et al. (2015). Chaos of rearrangements in the mating-type chromosomes of the anther-smut fungus Microbotryum lychnidis-dioicae. Genetics 200, 1275–1284. doi: 10.1534/genetics.115.177709
Bennett, J. W. (2005). Fungal secondary metabolism-from biochemistry to genomics. Nat. Rev. Microbiol. 3, 937–947. doi: 10.1038/nrmicro1286
Benson, G. (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bergmann, S., Schümann, J., Scherlach, K., Lange, C., Brakhage, A. A., and Hertweck, C. (2007). Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat. Chem. Biol. 3, 213–217. doi: 10.1038/nchembio869
Birney, E., Clamp, M., and Durbin, R. (2004). Genewise and genomewise. Genome Res. 14, 988–995. doi: 10.1101/gr.1865504
Blin, K., Wolf, T., Chevrette, M. G., Lu, X. W., Schwalen, C. J., Kautsar, S. A., et al. (2017). antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41.
Boraston, A. B., Bolam, D. N., Gilbert, H. J., and Davies, G. J. (2004). Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781. doi: 10.1042/bj20040892
Cantarel, B. L., Coutinho, P. M., Corinne, R., Thomas, B., Vincent, L., and Bernard, H. (2009). The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238.
Cheng, T. F., Jia, X. M., Ma, X. H., Lin, H. P., and Zhao, Y. H. (2004). Phylogenetic study on Shiraia bambusicola by rDNA sequence analyses. J. Basic Microb. 44, 339–350. doi: 10.1002/jobm.200410434
Chooi, Y. H., Zhang, G., Hu, J., Muria-Gonzalez, M. J., Tran, P. N., Pettitt, A., et al. (2017). Functional genomics-guided discovery of a light-activated phytotoxin in the wheat pathogen Parastagonospora nodorum via pathway activation. Environ. Microbiol. 19, 1975–1986. doi: 10.1111/1462-2920.13711
Daub, M. E., and Chung, K. R. (2009). Photoactivated perylenequinone toxins in plant pathogenesis. Plant Relationsh. 252, 197–206. doi: 10.1016/j.femsle.2005.08.033
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 113–132.
Faino, L., Seidl, M. F., Datema, E., Berg, G. C. M. V. D., Janssen, A., Wittenberg, A. H. J., et al. (2015). Single-molecule Real-Time sequencing combined with optical mapping yields completely finished fungal genome. mBio 6, 1–11.
Fischer, M., Knoll, M., Sirim, D., Wagner, F., Funke, S., and Pleiss, J. (2007). The cytochrome P450 engineering database: a navigation and prediction tool for the cytochrome P450 protein family. Bioinformatics 23, 2015–2017. doi: 10.1093/bioinformatics/btm268
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269.
Gao, L., Ma, Y. Y., Li, X. T., Zhang, L. P., Zhang, C., Chen, Q. Y., et al. (2019). Research on the roles of genes coding ATP-binding cassette transporters in Porphyromonas gingivalis pathogenicity. J Cell Biochem. 121, 93–102. doi: 10.1002/jcb.28887
Gardner, P. P., Daub, J., Tate, J. G., Nawrocki, E. P., Kolbe, D. L., Lindgreen, S., et al. (2009). Rfam: updates to the RNA families database. Nucleic Acids Res. 37, 136–140.
Gu, X. T., Zhou, J. H., Feng, Y. Y., Wei, S. H., Wang, X. S., and Zhang, B. W. (2005). Determination of Hypocrellin A in Hypocrella Bambusae by HPLC/DAD. J. Instru. Anal. 24, 130–132.
Guo, L. Y., Yan, S. Z., Li, Q., Xu, Q., Lin, X., Qi, S. S., et al. (2017). Poly(lactic-co-glycolic) acid nanoparticles improve oral bioavailability of hypocrellin A in rat. RSC Adv. 7, 42073–42082. doi: 10.1039/c7ra04748g
Hacquard, S., Kracher, B., Hiruma, K., Münch, P. C., Garrido-Oter, R., Thon, M. R., et al. (2016). Survival trade-offs in plant roots during colonization by closely related beneficial and pathogenic fungi. Nat. Commun. 7:11362.
Han, X., Chakrabortti, A., Zhu, J., Liang, Z. X., and Li, J. (2016). Sequencing and functional annotation of the whole genome of the filamentous fungus Aspergillus westerdijkiae. BMC Genomics 17:633. doi: 10.1186/s12864-016-2974-x
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, 286–293.
Jiang, H., Shen, Y., Liu, W., and Ling, L. (2014). Deletion of the putative stretch-activated ion channel Mid1 is hypervirulent in Aspergillus fumigatus. Fungal Genet Biol. 62, 62–70. doi: 10.1016/j.fgb.2013.11.003
Johnson, A. D., Handsaker, R. E., Pulit, S. L., Nizzari, M. M., O’Donnell, C. J., and Bakker, P. I. W. D. (2008). SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24, 2938–2939. doi: 10.1093/bioinformatics/btn564
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462.
Khalil, H. P. S. A., Bhat, I. U. H., Jawaid, M., Zaidon, A., Hermawan, D., and Hadi, Y. S. (2012). Bamboo fibre reinforced biocomposites: a review. Mater. Design. 42, 353–368. doi: 10.1016/j.matdes.2012.06.015
Kim, K. E., Peluso, P., Babayan, P., Yeadon, P. J., Yu, C., Fisher, W. W., et al. (2014). Long-read, whole-genome shotgun sequence data for five model organisms. Sci. Data 1, 140045–140055.
Kirk, P. M., Cannon, P. F., David, J. C., and Stalpers, J. A. (2008). Ainsworth and Bisby’s Dictionary of the Fungi, 10th Edn, Wallingford: CAB International.
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lagesen, K., Hallin, P., Rodland, E., Staerfeldt, H., Rognes, T., and Ussery, D. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lairson, L. L., Henrissat, B., Davies, G. J., and Withers, S. G. (2008). Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 77, 521–555. doi: 10.1146/annurev.biochem.76.061005.092322
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., Mcwilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Levasseur, A., Drula, E., Lombard, V., Coutinho, P. M., and Henrissat, B. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41–55.
Li, H., Coghlan, A., Ruan, J., Coin, L. J., Hériché, J. K., Osmotherly, L., et al. (2006). TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 34, D572–D580.
Lin, X., Yan, S. Z., Qi, S. S., Xu, Q., Han, S. S., Guo, L. Y., et al. (2017). Transferrin-modified nanoparticles for photodynamic therapy enhance the antitumor efficacy of hypocrellin A. Front. Pharmacol. 8:815. doi: 10.3389/fphar.2017.00815
Liu, B., Bao, J. Y., Zhang, Z. B., Yan, R. M., Wang, Y., Yang, H. L., et al. (2018). Enhanced production of perylenequinones in the endophytic fungus Shiraia sp. Slf14 by calcium/calmodulin signal transduction. Appl. Microbiol. Biot. 102, 153–163. doi: 10.1007/s00253-017-8602-0
Liu, Y. X., Hyde, K. D., Ariyawansa, H., Li, W. J., Zhou, D. Q., Yang, Y. L., et al. (2013). Shiraiaceae, new family of Pleosporales (Dothideomycetes, Ascomycota). Phytotaxa 103, 51–60.
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Archaeal clusters of orthologous genes (arCOGs): an update and application for analysis of shared features between Thermococcales, Methanococcales, and Methanobacteriales. Life 5, 818–840. doi: 10.3390/life5010818
Morakotkarn, D., Kawasaki, H., and Seki, T. (2007). Molecular diversity of bamboo-associated fungi isolated from Japan. FEMS Microbiol. Lett. 266, 10–19. doi: 10.1111/j.1574-6968.2006.00489.x
Nelson, D. R. (2011). Progress in tracing the evolutionary paths of cytochrome P450. BBA Proteins Proteom. 1814, 14–18. doi: 10.1016/j.bbapap.2010.08.008
Newman, A. G., and Townsend, C. A. (2016). Molecular characterization of the cercosporin biosynthetic pathway in the fungal plant pathogen Cercospora nicotianae. J. Am. Chem. Soc. 138, 4219–4228. doi: 10.1021/jacs.6b00633
O’Brien, E. M., Morgan, B. J., Mulrooney, C. A., Carroll, P. J., and Kozlowski, M. C. (2010). Perylenequinone natural products: total synthesis of hypocrellin A. J. Org. Chem. 75, 57–68. doi: 10.1021/jo901386d
Ohm, R. A., Nicolas, F., Bernard, H., Schoch, C. L., Horwitz, B. A., Barry, K. W., et al. (2012). Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 8:e1003037. doi: 10.1371/journal.ppat.1003037
Ospina-Giraldo, M. D., Griffith, J. G., Laird, E. W., and Mingora, C. (2010). The CAZyome of Phytophthora spp.: a comprehensive analysis of the gene complement coding for carbohydrate-active enzymes in species of the genus phytophthora. BMC Genomics 11:525. doi: 10.1186/1471-2164-11-525
Qi, S. S., Lin, X., Zhang, M. M., Yan, S. Z., Yu, S. Q., and Chen, S. L. (2014). Preparation and evaluation of hypocrellin A loaded poly (lactic-co-glycolic acid) nanoparticles for photodynamic therapy. RSC Adv. 4, 40085–40094. doi: 10.1039/c4ra05796a
Robertson, C. A., Evans, D. H., and Abrahamse, H. (2009). Photodynamic therapy (PDT): a short review on cellular mechanisms and cancer research applications for PDT. J. Photoch. Photobio. B 96, 1–8. doi: 10.1016/j.jphotobiol.2009.04.001
Rogers, S. O., and Bendich, A. J. (1994). “Extraction of total cellular DNA from plants, algae and fungi,” in Plant Molecular Biology Manual, eds S. B. Gelvin and R. A. Schilperoort (Dordrecht: Springer), 183–190. doi: 10.1007/978-94-011-0511-8_12
Rokas, A., Williams, B. L., Nicole, K., and Carroll, S. B. (2003). Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425, 798–804. doi: 10.1038/nature02053
Ruan, J., Li, H., Chen, Z. Z., Coghlan, A., Coin, L. J., Guo, Y., et al. (2008). Treefam: 2008 update. Nucleic Acids Res. 36, D735–D740.
Rytioja, J., Hildén, K., Yuzon, J., Hatakka, A., Vries, R. P. D., and Mäkelä, M. R. (2014). Plant-polysaccharide-degrading enzymes from Basidiomycetes. Microbiol. Mol. Biol. R. 78, 614–649. doi: 10.1128/mmbr.00035-14
Shen, X. Y., Li, T., Chen, S., Fan, L., Gao, J., and Hou, C. L. (2015). Characterization and phylogenetic analysis of the mitochondrial genome of Shiraia bambusicola reveals special features in the order of pleosporales. PLoS One 10:0116466. doi: 10.1371/journal.pone.0116466
Sit, C. S., Ruzzini, A. C., Van Arnam, E. B., Ramadhar, T. R., Currie, C. R., and Clardy, J. (2015). Variable genetic architectures produce virtually identical molecules in bacterial symbionts of fungus-growing ants. Proc. Natl. Acad. Sci. U.S.A. 112, 13150–13154. doi: 10.1073/pnas.1515348112
Stanke, M., Diekhans, M., Baertsch, R., and Haussler, D. (2008). Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644. doi: 10.1093/bioinformatics/btn013
Ter-Hovhannisyan, V., Lomsadze, A., Chernoff, Y. O., and Borodovsky, M. (2008). Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 18, 1979–1990. doi: 10.1101/gr.081612.108
Thieme, K. G., Jennifer, G., Sasse, C., Valerius, O., Thieme, S., Karimi, R., et al. (2018). Velvet domain protein VosA represses the zinc cluster transcription factor SclB regulatory network for Aspergillus nidulans asexual development, oxidative stress response and secondary metabolism. PLoS Genet. 14:e1007511. doi: 10.1371/journal.pgen.1007511
Torto-Alalibo, T., Collmer, C. W., and Gwinn-Giglio, M. (2009). The plant-associated microbe gene ontology (PAMGO) consortium: community development of new gene ontology terms describing biological processes involved in microbe-host interactions. BMC Microbiol. 9:S1. doi: 10.1186/1471-2180-9-S1-S1
Tsuji, M., Kudoh, S., and Hoshino, T. (2015). Draft genome sequence of cryophilic basidiomycetous yeast Mrakia blollopis SK-4, isolated from an algal mat of Naga-ike lake in the Skarvsnes ice-free area, east Antarctica. Genome Announc. 3:e1454.
Wang, Q. H., Chen, D. P., Wu, M. C., Zhu, J. D., Jiang, C., Xu, J. R., et al. (2018). MFS transporters and GABA metabolism are involved in the Self-defense against DON in Fusarium graminearum. Front. Plant Sci. 9:438. doi: 10.3389/fpls.2018.00438
Yong, Z., Kang, Z., Fang, A., Han, Y., Yang, J., Xue, M., et al. (2014). Specific adaptation of Ustilaginoidea virens in occupying host florets revealed by comparative and functional genomics. Nat. Commun. 5, 1–12.
Zhao, N., Lin, X., Qi, S. S., Luo, Z. M., and Chen, S. L. (2016). De novo transcriptome assembly in Shiraia bambusicola to investigate putative genes involved in the biosynthesis of hypocrellin A. Int. J. Mol. Sci. 17, 311–324.
Keywords: Shiraia bambusicola, pathogenicity, phylogeny, secondary metabolites, biosynthesis
Citation: Zhao N, Li D, Guo B-J, Tao X, Lin X, Yan S-Z and Chen S-L (2020) Genome Sequencing and Analysis of the Hypocrellin-Producing Fungus Shiraia bambusicola S4201. Front. Microbiol. 11:643. doi: 10.3389/fmicb.2020.00643
Received: 28 April 2019; Accepted: 20 March 2020;
Published: 09 April 2020.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Kathryn Bushley, University of Minnesota Twin Cities, United StatesMichael H. Perlin, University of Louisville, United States
Copyright © 2020 Zhao, Li, Guo, Tao, Lin, Yan and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-Zhen Yan, yanshuzhen@njnu.edu.cn; Shuang-Lin Chen, chenshuanglin@njnu.edu.cn; nnucylab@126.com