 Lichun Ye1†
Lichun Ye1† Lulu Yang2
Lulu Yang2 Bo Wang3Gang Chen2Liping Jiang4
Bo Wang3Gang Chen2Liping Jiang4 Zhigang Hu1
Zhigang Hu1 Zhaohua Shi5*
Zhaohua Shi5* Yifei Liu1*†
Yifei Liu1*† Shilin Chen1,6,7*
Shilin Chen1,6,7*- 1College of Pharmacy, Hubei University of Chinese Medicine, Wuhan, China
- 2Genomics Project Department, Wuhan Benagen Tech Solutions Company Limited, Wuhan, China
- 3Hubei Institute for Drug Control, Wuhan, China
- 4Department of Pharmacy, Wuhan Hospital of Traditional and Western Medicine, Wuhan, China
- 5Key Laboratory of Chinese Medicine Resource and Compound Prescription, Ministry of Education, Hubei University of Chinese Medicine, Wuhan, China
- 6Institute of herbgenomics, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 7Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, China
Aesculus L. (buckeye and horse chestnut) are woody plant species with important horticultural and medicinal values. Aesculus seeds are widely used as biomedicine and cosmetic ingredients due to their saponins. We report a chromosomal-scale genome of Aesculus wilsonii. Sequences amounting to a total of 579.01 Mb were assembled into 20 chromosomes. More than half of the genome (54.46%) were annotated as repetitive sequences, and 46,914 protein-coding genes were predicted. In addition to the widespread gamma event with core eudicots, a unique whole-genome duplication (WGD) event (17.69 Mya) occurred in Aesculus after buckeye differentiated from longan. Due to WGD events and tandem duplications, the related synthetic genes of triterpene saponins unique to Aesculus increased significantly. Combined with transcriptome characterization, the study preliminarily resolved the biosynthetic pathway of triterpenoid saponins like aescin in A. wilsonii genome. Analyses of the resequencing of 104 buckeye accessions revealed clear relationship between the geographic distribution and genetic differentiation of buckeye trees in China. We found that the buckeye species found in southern Shaanxi is A. wilsonii rather than A. chinensis. Population dynamics analysis further suggests that the population size and evolution of existing buckeye species have been influenced by climate fluctuations during the Pleistocene and recent domestication events. The genome of A. wilsonii and population genomics of Aesculus provide a resource for future research on Hippocastanaceae. These findings will contribute to the utilization and diversity protection of Aesculus.
Introduction
Buckeye trees (Aesculus, Hippocastanaceae, Sapindales) are important medicinal and ornamental plants (Barton and Castle, 1877). In the Pharmacopoeia of the People’s Republic of China (2020 ed.), the seeds of A. wilsonii, A. chinensis, and A. chekiangensis are reported to be sources of traditional Chinese medicine (TCM) such as Semen Aesculi (Suo Luo Zi), which is used for treating dyspnea, abdominal distention, and epigastralgia (Commission, C.P, 2020). In Europe, the seeds and the bark of young branches of Aesculus hippocastanum (horse chestnut) are also widely used as medicines (Braga et al., 2012; Idris et al., 2020). The therapeutic properties of Aesculus seeds were first recorded in Zhou Hou Bei Ji Fang, which was written by Ge Hong during the Eastern Jin Dynasty in China to treat abdominal pain and for detoxification (Ge, 1963). In the Compendium of Materia Medica (Bencao Gangmu), Aesculus seeds were described as sweet-warm and non-toxic, and recommended for treatment of rheumatic hands and foot spasms (Li, 1982). The chemicals of Semen Aesculi include triterpenoid saponins, flavonoids, and lipids. The aescins with the oleanane type triterpenoid saponins are the main active ingredients in seed (Zhang et al., 2010), with anti-ischemic edema, anti-inflammatory, and anti-tumor pharmacological properties (Cheong et al., 2018; Gallelli, 2019).
Triterpene saponins are widely present in the normal growth and development of various plants. Previous studies have paid wide attention on the biosynthesis of triterpenoid saponins in plants (Thimmappa et al., 2014). Triterpenoid saponins are typically synthesized through the isoprenoid pathway, and cyclized by of 2,3-oxidosqualene to give oleanane (β-amyrin) or dammarane triterpenoid skeletons (Haralampidis et al., 2002). A variety of oxidosqualene cyclases (OSCs) are charge for the cyclization step, which marks the branch point in the metabolism of primary and secondary triterpenes. The triterpenoid backbones are then modified by various cytochrome P450-dependent monooxygenases (CYPs), UDP-glycosyltransferases (UGTs), acyltransferases, methyltransferases, and others to increase the complexity, expand the structure and functional diversity (Thimmappa et al., 2014). However, knowledge of the relationships of enzymes and biochemical pathways involved in saponin biosynthesis remains limited. Novel functionalization of a cellulose synthase-like (Csl) enzyme with a form of triterpenoid glycosyltransferase activity enables glucuronic acid attach to the C-3 position of saponins like aescins (Jozwiak et al., 2020). The major aescins (aescin1a, aescin1b, isoescin 1a, isoescin 1b) in the seeds are the parent nucleus of oleanolic acid with three glucose-forming glycoside substitutions at the C3 position. The subtle different between aescins were substituted by cis-trans ethylenic configuration at C21 position, and whether the C22 and C28 positions were substituted by acetyl (Colson et al., 2019). Little has been reported about the biosynthetic pathway of aescins due to the lack of reliable genomic information.
Approximately 12–19 species of Aesculus have been identified in eastern Asia, eastern and western North America, and southeastern Europe (Hardin, 1957a; Hardin, 1957b; Hardin, 1957c). Based on morphological and DNA sequence data from ITS, rbcL, and matK, all Aesculus species represent a monophyletic group that can be further divided into five sections. The Calothyrsus section includes species that mainly occur in eastern Asia (Xiang et al., 1998; Harris and Thomas, 2009; Du et al., 2020). Three TCM species (A. wilsonii, A. chinensis, and A. chekiangensis) and Aesculus wangii from Yunnan are relatively widespread taxa in this section. They are closely related and have very similar morphologies (FANG, 1981). For example, cultivated buckeye plants from southern Jiangsu and northern Zhejiang are described as A. chekiangensis, which has been considered to be a variety of A. chinensis due to its very similar morphology (Xia and Turland, 2005). Introgression or hybridization is also possible given that numerous intermediates between A. wilsonii and A. chinensis occur within the natural range of A. wilsonii (Hardin, 1957c). Clarifying the genetic relationship of buckeye species in China is therefore important for the optimization of their medicinal applications.
In this study, we sequenced and assembled a chromosome-scale genome of A. wilsonii (Figures 1A–E). We performed a phylogenetic and comparative genome analysis to investigate the phylogenetic position of A. wilsonii within representative flowering plants and also the ancient whole-genome duplication (WGD) events occurring during A. wilsonii evolution. With available genome data, the main gene families and genes related in the biosynthesis of triterpenoid saponin were also examined, and their expression in different tissues were documented. Resequencing of population samples of A. wilsonii and A. chinensis, A. chekiangensis, and A. wangii in China were also conducted to clarify their genetic relationships and evolutionary demography. The results provide a reference for the resource conservation and further utilization of Aesculus plants.

Figure 1 Morphological characteristics of Aesculus wilsonii. (A) A mature A. wilsonii tree growing in Shennongjia National Nature Reserve. (B) Mature leaves of A. wilsonii. (C, D) Flowers. (E) Fruits and seed, Bar=1cm.
Materials and methods
Plant materials
The A. wilsonii tree used for genome sequencing was from Shennongjia National Nature Reserve, Hubei, China. Fresh young leaves were collected and immediately frozen for next DNA extraction. A total of 104 resequencing Aesculus samples were collected from 17 populations from six provinces, representing four different species including A. wilsonii, A. chinensis, A. chekiangensis, and A. wangii (Supplementary Table 1).
Genome library construction and sequencing
By the modified CTAB method (Porebski et al., 1997), we extracted high-quality genomic DNA from young leaves, and checked the quality and quantity of the isolated DNA by A260/A280 nm and 0.5% agarose gel electrophoresis. A paired-end library with short-insert (300–350 base pairs) was constructed following manufacturer protocol (Illumina). To ensure the library quality, Agilent 2100 and q-PCR were used to detect the inserted fragments, and accurately quantify the effective concentration of the library. Quality-compliant libraries were sequenced using the Illumina NovaSeq platform. The genomic DNA was purified and fractionated by the BluePippin system (Sage Science) and then used to construct the sequencing libraries by the protocol provided with the SQK-LSK109 genomic sequencing kit (Oxford Nanopore Technologies) for the Oxford Nanopore MinION platform (ONT).
Hi-C sequencing
The DNA of young leaves was extracted after fixing chromatin with formaldehyde for Hi-C sequencing. Qualified samples were digested by restriction endonuclease. The fragments were purified after labeling the biotin nucleotides and ligating the blunt ends to each other. The Hi-C fragment was processed by removal of biotin, ultrasound interruption, terminal repair, addition of base A and PCR amplification to construct the library. A 150-bp paired-end mode was used to sequence the library on an Illumina NovaSeq System.
RNA sequencing
The total RNA of the flowers, leaves, seeds of A. wilsonii were extracted using the R6827 Plant RNA Kit following manufacturer instructions. After the RNA samples were qualified, the entire library was prepared by random interruption, end repair, A-tail addition, sequencing adapter addition, purification, and PCR amplification. After the libraries were qualified, according to the requirements of effective concentration and target data volume, the different libraries were pooled to the Flow cell and sequenced by the Illumina NovaSeq platform. The raw data was filtered by fastp (ver.: 0.21.0; parameter: default) to obtain clean data, and we aligned the filtered transcriptome sequence with the reference genome according to Star (version: 2.7.9a; parameter: default). We aligned the number of reads of each transcript for each sample by RSEM, and performed fragments per kilobase per million bases (FPKM) calculation to obtain the expression levels of genes and transcripts.
Genome survey and assembly
The genome size and heterozygosity of A. wilsonii were estimated with short reads by k-mer analysis. With GCE and Genome Scope tool (Liu et al., 2013), the genome size was estimated by dividing the total number of k-mers counted by the k-mer coverage based on the 19-mer frequency of Illumina short reads, and the first estimate of the GC content, heterozygosity, and repeat content. The clean long reads were filtered by MinKNOW from the raw fastq data of ONT sequencing were corrected and assembled by SMART de novo (Schmidt et al., 2017) with default parameters. Racon (Vaser et al., 2017) (Version:1.4.11) software was used to conducted two rounds of polishing based on third-generation sequencing data, and two rounds were polished with Pilon (Walker et al., 2014)(version:1.23) by Illumina short reads. The map_rate with Illumina reads, coverage were used to estimate the quality of the genome assembly by the BUSCO (Simão et al., 2015) (v.3.0.1). The Hi-C clean data were aligned to the preceding scaffold assembly by using BWA software with default parameters (Li and Durbin, 2009). SAMTOOLS (Li et al., 2009) was used to remove duplicates (parameters: rmdup) and nonaligned data. Statistical analysis was performed on unique mapped reads, and unique read pairs with interaction effectively around the restriction site were identified to be used to build the pseudo-chromosome sequences. ALLHiC (Zhang et al., 2019) (v0.9.12) was used to construct the scaffold graph by optimizing the ordering and orientation of each clustered group within simple diploid mode. Finally, the scaffolds were anchored to 20 chromosomes, and an interaction heat map of each chromosome heatmap was plotted.
Genome annotation
We combined the de novo and homology-based methods to identify the repeat sequences. The repetitive sequences in the genome of A. wilsonii were filtered by RepeatModeler (Flynn et al., 2020) (version:1.0.4) to constructed the repeat library. Then we combed the repeat library and the repbase-derived RepeatMasker library. Based on homology in RepeatMasker (Chen, 2004), the repetitive sequences were identified by a conserved BLASTN search. Non-coding RNAs can also have important biological functions. Based on their structural characteristics, tRNAsan-SE (Lowe and Eddy, 1997)(version1.23) was used to search the tRNAs in the A. wilsonii genome, and RNAmmer (version: 1.2) was used to predict the rRNA. According INFERNAL (Nawrocki and Eddy, 2013)(Version: 1.1.2), the ncRNA and snRNAs sequences in the genome were identified based on the Rfam (Ioanna et al., 2018) database. We used MAKER (Kondrashov et al., 2011)(version:2.31.8) combined de novo and homology and transcriptome-based predictions, to predict protein-coding genes. The transcriptome results were de novo spliced by Trinity (Grabherr et al., 2013) (version: v2.6.6) and annotated as EST data. We used the model of Augustus (Hoff et al., 2016) (tomato) combined with other species including Solanum lycopersicum, Arabidopsis thaliana, Vitis vinifera, Oryza sativa, Dimocarpus longan, Citrus sinensis, Atalantia buxifolia, Citrus reticulata, Citrus grandis and Helianthus annuus for genetic structure prediction. The functional annotation of all A. wilsonii genes was performed by homologous alignments with BLASTP (version:0.7.9,version,e-value ≤1e−5) in public protein databases such as Uniprot (https://www.uniprot.org/), NR, KEGG (Minoru et al., 2014), InterProScan (Zdobnov and Rolf, 2001) (version: 5.33–72.0), and Pfam (Finn et al., 2008).
Genome evolution
Ten other species—A. buxifolia, D. longan, C. reticulata, C. grandis, C. sinensis, C. nankingense, H. annuus, A. thaliana, V. vinifera, and O. sativa—were selected for identifying the gene family of A. wilsonii by OrthoMCL (Li et al., 2003). We aligned 1521 single-copy genes that were common to the 11 selected genomes using MUSCLE (Edgar, 2004) (version: V3.8.31). alignment results were filtered (-GT 0.2) by trimal (version: V1.4.rev 22). Then the phylogenetic tree with PROTGAMMAWAG model was constructed by RAxML (Kevin et al., 2011) (Version: 8.2.10). CAFÉ (Bie et al., 2006) (version: 2.1; Parameter: – filter) was used for gene family contraction and expansion analysis. Using referenced the fossil nodes were selected from TimeTree (Sudhir et al., 2017) (http://www.timetree.org/), MCMCtree was applied to estimate the divergence time of 11 species.in PAML (Yang, 2007) (version 4.9). The Ka/Ks ratios of genes calculated by the PAML package were used to detect the positive selection genes with a threshold value of Ka/Ks ≥ 1 and a P value ≤.05.
Synteny and genomic duplication analysis
Whole-genome duplication (WGD) events are important for the adaptive genome evolution of species. The protein sequences of different species were matched using BLAST (version: 2.6.0+, parameters: e-value 1–5 - e outfmt 6), then MCScanX (Tang et al., 2008) (https://github.com/wyp1125/MCScanx; Parameters: -a - E 1E-5-S 5) were used to analyze collinear blocks of the intra- and interspecies comparisons of the A. wilsonii genome. PAML (version: 4.9, yn00) was used to calculate the synonymous mutation frequency (Ks), non-synonymous mutation frequency (Ka), and the ratio of non-synonymous mutation rate to synonymous mutation rate (Ka/Ks) of collinear gene pairs, and they were plotted using GGploT2 (version: 2.2.1). According to T=Ks/2r, the divergence time was converted by calculated Ks value with a substitution rate in eudicots was 6.5×10-9 mutations per site per year.
Genes related to pentacyclic triterpenoid saponins biosynthesis
Pentacyclic triterpenoid saponins are the main medicinal component of A. wilsonii. We retrieved protein sequences of the terpenoid backbone, sesquiterpenoid and triterpenoid biosynthesis pathway (00900,00909) from Glycine max and Arabidopsis thaliana, including ACAT, HMGS, HMGR, MVK, PMK, MVD, IDI, DXS, DXR, MCT, CMK, MDS, HDS, HDR, FPPS, GPPS, GPS, SQS, and SQE from the NCBI database. Based on these homologs as queries, we identified the candidate protein sequences for homologs to these proteins in the A. wilsonii genome. The OSC, CYP450, BAHD, SCPL, Csl, and UGT genes were predicted using hmmsearch according the OSC hmm model (PF13249 and PF13243), the CYP450 hmm model (PF00067), BAHD hmm model (PF02458), SCPL hmm model (PF00450), Csl hmm model (PF03552), and UGT hmm model (PF00201) from Pfam and homolog-based BLAST with an e-value cutoff of 1e−5. Using Mega-X (Kumar et al., 2018) (V10.0.5), phylogenetic trees of each gene families related biosynthesis of triterpenoid saponins were constructed from A. thaliana and A. wilsonii. Then according to the reported reference, the predicted OSC, CYP450, BAHD, SCPL, Csl, and UGT candidates were further divided into clans or families with similar functions.
Resequencing and quality control
DNAs from leaf tissue of 104 buckeye accessions were isolated by the modified CTAB method. After control of the quality and quantity by 0.5% agarose gel electrophoresis and A260/A280 nm, 104 DNA samples were used for the construction of the libraries on the Illumina NovaSeq 6000 with an expected target coverage of 5×. In total, 369.33 Gb of clean bases were obtained. The clean reads were aligned to the reference genome by BWA (Li and Durbin, 2009)(version:0.7.17, Parameters: mem To obtain high quality SNP, GATK (version: 4.2.0.0, parameter: VariantFiltration) was used to filter the SNP following the officially recommended standard (QD < 2.0 || QUAL < 30.0 || SOR > 3.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0).
Analysis of population structure
Based on the results following GATK hard-filtering, vcftool (Danecek et al., 2011) (version: 0.1.16, parameters: – maf, – max-missing) was used to eliminate minor allele frequencies less than 0.05. The final SNPs of 104 samples were performed to construct a neighbor-joining phylogenetic tree by PHYLIP (Purcell et al., 2007) (version 3.696), and implemented visualization within ggtree (Yu, 2020) based on newick tree file. To illustrate the population genetic structure of buckeye, ADMIXTURE (version: 1.3.0; -cv input File K) was used to estimate the optimal ancestral population from a multi-locus SNP genotype dataset. When the number of subgroups contained in the population was not known, the range of K can be preset from 2 to N. Through a simulation of calculation, the classification of the population and the proportion of each sample’s pedigree can be calculated based on the Bayesian algorithm. We selected the best K value according to error value and maximum likelihood value (Alexander et al., 2009). Based on the degree of variation difference among population samples, GCTA (Yang et al., 2011)(version: 1.93.2, parameters: -GRM, -PCA) was used to cluster individuals into different subgroups according to the PCA method. Based on the probability of simultaneous occurrence of two or more alleles at loci, PopLDdecay (version: 3.26) was used to calculate the R2 of the maximum distances of 300 Kb by genome-wide linkage disequilibrium analysis (Vos et al., 2017). The genetic differentiation (FST), nucleotide diversity (π), and relatedness PHI were calculated by Vcftools (version: 0.1.16) to analyze the relationships between different subgroups.
Gene flow and demographic history analysis
To infer the patterns of differentiation and mixing in multiple populations, Treemix (Pickrell et al., 2012) was used to evaluate the gene flow between populations. Based on the genome-wide alleles set, the actual covariance (Real value) between each pair of populations was calculated, and the maximum likelihood trees were constructed to calculate the estimated value of covariance. The difference between the actual and estimated value was used to judge whether gene flow occurs between the two populations. The demographic history of each Aesculus population were investigated by the PSMC (Schiffels and Durbin, 2014) with the mutation rate 1.722×10−9 per base per generation (Xiang et al., 1998) and the generation time g = 8 years over the last 20 million years.
Results
Genome sequencing and assembly
A. wilsonii is a diploid (2n = 2× = 40), with the genome size was estimated to be 552.87 Mb according to k-mer (k = 19) analysis and a relatively high genome heterozygosity (1.22%) (Supplementary Table 2; Supplementary Figure 1). The estimated genome size of A. wilsonii is very close to those previously reported for other Aesculus taxa ranging from 467 to 623.48 Mb (Krahulcová et al., 2017). According Nanopore genome sequencing, a total of 52.11 Gb of raw long reads (Supplementary Table 3) were produced. After being filtered, the clean long reads were further self-corrected and polished with 63.76 Gb short reads, and finally assembled to produce a primary genome of 611.22 Mb, including 376 sequence contigs (N50 = 3.75 Mb) (Supplementary Table 4).
In order to assemble into the chromosome-scale genome of A. wilsonii, 221,213,230 read pairs were obtained by Hi-C sequencing, of which 94.72% of reads were mapped to the assembled contigs, including 75,353,070 uniquely mapped read pairs and 29,283,408 read pairs that represented valid interactions (Supplementary Table 5). All the mapped contigs were categorized and ordered to construct chromosome-scale scaffolds. Finally, 94.72% scaffolds were anchored to 20 pseudochromosomes (Figure 2A; Supplementary Figure 2), resulting in a chromosome-level A. wilsonii genome assembly of 579.01 Mb (scaffold N50 = 28.02 Mb) (Supplementary Tables 4, 6). Further analysis by mapping Illumina short reads showed that about 98.79% reads could be mapped back to the genome assembly (Supplementary Table 7). All these collectively suggested that the A. wilsonii genome assembly with high quality.
Genome annotation and repetitive elements
An integrated strategy including the ab initio gene prediction and evidence-based methods were used to annotate the protein-coding of the A. wilsonii genome (Supplementary Figure 3). After removing nonfunctional annotations, 46,914 protein-coding genes was retained (Supplementary Table 8). It was a near completeness of gene prediction with 91% annotated BUSCO gene models were identified by BUSCO software (Supplementary Table 9). The 46,914 genes were identified, and 44210 proteins (94.24%) were annotated using four databases of known proteins (Supplementary Table 10). In addition, 4282 noncoding RNAs were identified in the A. wilsonii genome, including 302 microRNAs, 801 transfer RNAs, 1866 ribosomal RNAs, and 1225 small nuclear RNAs (Supplementary Table 11). Approximately 54.46% (332.85 Mb) of the repetitive elements were present in the genome of A. wilsonii (Figure 2A and Supplementary Table 12). Among these, long terminal repeat (LTR) retrotransposons, DNA transposable elements, long interspersed nuclear elements (LINE) accounted for 26.99%, 7.20%, and 2.99%, respectively. Both Gypsy (16.17%) and Copia (10.05%) LTRs were widely present in the A. wilsonii genome.
Genome Evolution
To explore the evolutionary relationship between A. wilsonii and other plant species, we collected the data of 10 sequenced plant genomes, including genomes from five species of Sapindaceae (Atalantia buxifolia, Citrus reticulate, Citrus sinensis, Citrus grandis, and Dimocarpus longan) to construct a phylogenetic tree with 1,521 single copy ortholog genes shared from these genomes. (Supplementary Table 13). The resulting phylogenetic tree showed a sister relationship between A. wilsonii and D. longan, both of which are in the Sapindaceae. All of the Sapindaceae species were clustered into one monophyletic group. Using MCMCtree with calibrations, A. wilsonii and D. longan diverged from the last common ancestor at ~114.2 Mya, the divergence time between A. wilsonii and the most recent common ancestor of other Sapindaceae species at approximately 152.3 Mya (Figure 2B).
By comparing these genomes, a total of 18,267 orthologous genes were identified, including 2,090 genes unique to A. wilsonii (Supplementary Figure 4; Supplementary Table 13). In addition, evolution analysis of gene families revealed that 2,847 gene families in A. wilsonii expanded, whereas 771 families contracted (Figure 2B). Among the significantly expanded, or unique, gene families of A. wilsonii, functional annotation revealed that they were mainly enriched in Gene Ontology (GO) functional categories or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, such as energy metabolism and transport, DNA reparation, glucose metabolism, hormonal signal transduction, biosynthesis of aromatic compounds, and flavonoid biosynthesis (Supplementary Figures 5, 6). Analysis of the Ka/Ks ratio of A. wilsonii, D. longan, C. sinensis, A. buxifolia, and Vitis vinifera also indicated that A. wilsonii had a moderate rate of gene evolution (Supplementary Figure 7).
Whole-genome duplication of A. wilsonii
In order to explore whole-genome duplication events during A. wilsonii evolution, the age distribution of the duplication events was calculated by using the Ks values of the duplicated genes. Two main distribution peaks of A. wilsonii were found around the Ks values of 0.23 and 1.42 (Figure 2C). Ks 1.42 was consistent with the ancestral palaeohexaploidy event (γ event), which was shared by most core eudicots. The Ks 0.23 peak (∼17.69 Mya) suggested that a recent WGD event occurred in A. wilsonii. The same analysis with the genomes of D. longan, C. sinensis, A. buxifolia, and V. vinifera revealed that an occurrence of recent WGD event in A. wilsonii was lineage-specific and distinct from WGD events identified in other species (Figure 2C). Two Ks peak values at 0.3 and 0.64 were presented by comparing the homologous genes between A. wilsonii and D. longan, and between A. wilsonii and C. sinensis, respectively (Figure 2C). The result indicated that the recent WGD event in A. wilsonii occurred after the divergence of A. wilsonii from both C. sinensis and D. longan.
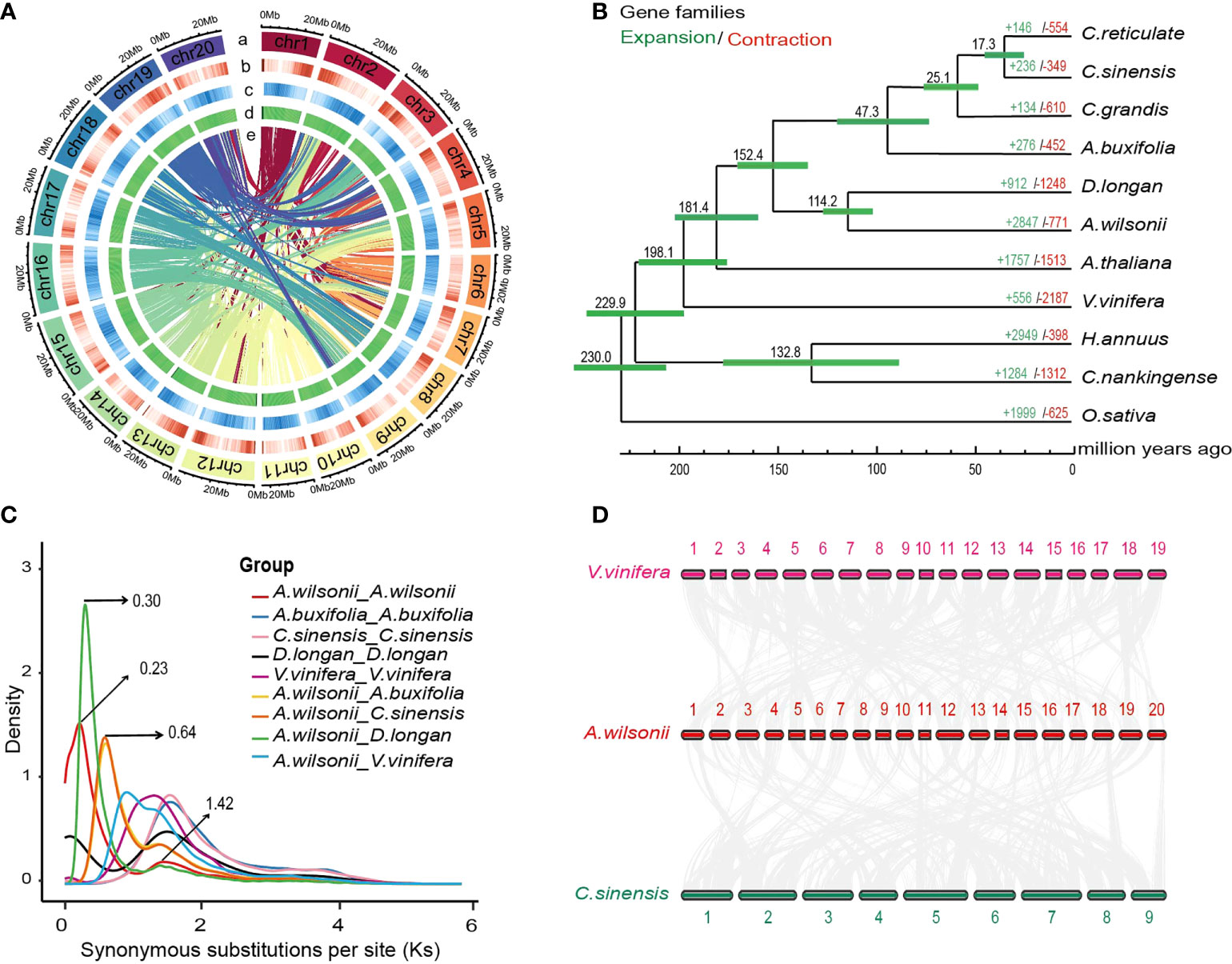
Figure 2 Genome evolution of Aesculus wilsonii. (A) Aesculus wilsonii genomic landscape. a Assembled pseudochromosome, b Gene density, c Repeat density, d GC content, e Syntenic blocks colored lines (from outside to inside). (B) The phylogenetic relationships and evolution analysis of A. wilsonii with other species. Green and red fonts indicate the numbers of expanded and contracted gene families that in 11 species, respectively. The green bars represent the 95% confidence interval of the estimated divergence time. (C) Ks distribution of syntenic blocks between A. wilsonii, A. buxifolia, C. sinensis, D. longan and V. vinifera. (D) Identified synteny blocks between A. wilsonii, V. vinifera and C. sinensis.
To confirm the recent WGD event presented in A. wilsonii, we performed a comparison of the syntenic depth ratio between A. wilsonii and the V. vinifera and C. sinensis genomes. We observed a similar pattern of syntenic depth ratio of four-to-two between A. wilsonii and V. vinifera, as well as between A. wilsonii and C. sinensis (Supplementary Figure 8). This indicates that a single syntenic region in both V. vinifera and C. sinensis is aligned to two A. wilsonii blocks (Figure 2D; Supplementary Figure 9). The results of the intergenomic co-linearity analysis provided clear support for the presence of a lineage-specific WGD event in A. wilsonii.
Evolution of genes related to triterpenoid saponins
Aescins in A. wilsonii are the pentacyclic triterpene saponins of the oleanane type (Gruza et al., 2013), which are mainly produced through the mevalonate (MVA) and 2-c-methyl-d-erythritol-4-phosphate (MEP) metabolic pathways (Figure 3A). Isopentenyl diphosphate (IPP) is synthesized from pyruvate and glyceraldehyde-3-phosphate through seven enzyme reactions including DXS, DXR, MCT, CMK, MDS, HDS, HDR in plastids, and IPP is transported to the cytoplasm. In addition, acetyl CoA is the initial donor for producing dimethylallyl pyrophosphate (DMAPP) through the six enzymes AACT, HMGS, HMGR, MK, PMK, MVD in the cytoplasm. IPP and DMAPP can be converted to each other in the cytoplasm under the action of isopentenyl diphosphate-isomerase (IDI) (Pu et al., 2021) (Supplementary Figure 10). Under the catalysis of FPPS, GPPS, GPS, SQS, SQE, squalene 2,3-epoxide is formed from IPP and DMAPP as precursors (Supplementary Figure 11). The squalene 2,3-epoxide catalyzed by β-Amyrin Synthase (β-AS) in the OSC family to produce amyrin. The amyrin is further modified by triterpene-modifying (or tailoring) enzymes such as cytochrome P450s (CYP450), sugar transferases (UGTs), and acyltransferases (ACTs) to form a variety of triterpenoids (Thimmappa et al., 2014). According to the annotation and homology comparison with other species, the results show that genes in triterpene saponin pathway exist widely in the genome of A. wilsonii (Supplementary Figure 12).
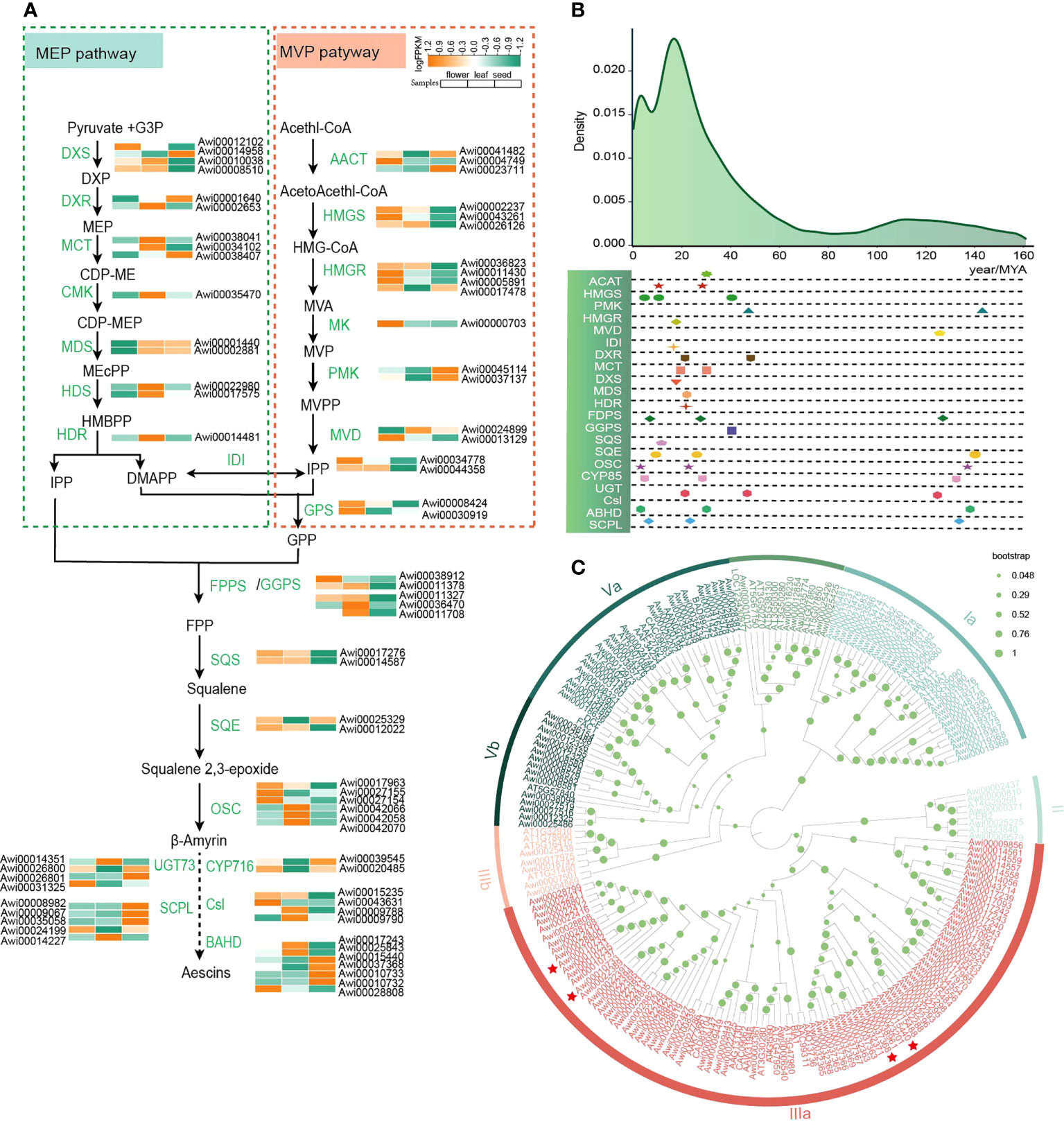
Figure 3 Evolution of genes involved in triterpenoid saponins in A. wilsonii. (A) Metabolic pathways and genes expression associated with biosynthesis and accumulation of triterpenoid saponins in A. wilsonii (B) The evolution of genes involved in triterpenoid saponins synthesis in A. wilsonii. (C) A phylogenetic tree of the BAHD genes in A. wilsonii. Among the seven branches, IIIA mostly uses acetyl-CoA as the main acyl donor, and accepts different alcohols as substrates to form esters. Genes marked with red stars were significantly expressed in seeds.
The OSC family includes important enzymes involved in the formation of triterpenes. OSC catalyzes squalene 2,3-epoxide to form more than 100 triterpenes. β-AS is the only one that can catalyze the formation of oleanane type triterpenoid saponins (Xu et al., 2004). We identified 19 OSC genes. Three of the 19 OSC genes were β-ASs and 10 synthases with integrative functions (Supplementary Figure13). Genes encoding specific metabolic pathways are physically aggregated in plant genome genomes and often co induced (Nutzmann et al., 2016; Nutzmann et al., 2018). According to the research of OSC flanking region genes in different Brassicaceae, the core genes encoding triterpenes containing OSCs, CYP450s, and ACTs to form the dynamic gene clusters, diversified the enzymes to cope with evolutionary selection pressures (Liu et al., 2020a). In the A. wilsonii genome, we identified a total of 320 CYP450s (Supplementary Figure 14), and eight genes belonged to the CYP716 family and were reported to catalyze the C28 position in other plants (Nelson and Werck-Reichhart, 2011; Zhang et al., 2020). In addition, we identified 153 genes from the BAHD family (D'Auria, 2006; Johnson et al., 2011) and 46 genes from the serine carboxypeptidase-like (SCPL) acyltransferase family (Fraser et al., 2005). The members of BAHD family were divided into 7 classes, including Ia, Ib, II, IIIa, IIIb, Va, and Vb. The SCPL family was divided into the IA, IB, II, III, IV, V, VI classes (Figure 3C; Supplementary Figure 15). Among them, branch III in BAHD was reported to form esters with acetyl-CoA as the main acyl donor and various alcohols as substrates, and IA in the SCPL family has an acylation function.
We classified 230 members of the UGT family into 23 clans (Supplementary Figure 16). The glycosides of the aescin mainly consist of different trisaccharide chains from xylose, galactose, and glucose, and form glycoside bonds with C3-OH through glucuronic acid (Augustin et al., 2012). We identified 38 genes of the UGT73 family involved in glucuronic acid transfer in the genome of A. wilsonii. In addition, a total of 25 cellulose synthase-like genes were identified, of which 7 genes in clade Csl M were reported to enable glucuronic acid attach to the C-3 position during saponin synthesis (Supplementary Figure 17). The expression of these genes related to the triterpenoid saponins biosynthesis among various tissues showed that the genes related to terpenoid backbone biosynthesis (KO00900) and triterpenoid synthesis (KO00909) are abundantly expressed in A. wilsonii (Figure 3A). Especially in the late stage of saponin biosynthesis, the expression profiles showed that genes related to aescin biosynthesis such as Awi00026800 and Awi00026801 in the UGT73 family; Awi00039545 and Awi00020485 in CYP716; Awi00015440, Awi00037368, Awi00010733, and Awi00010732 in the IIIa clade of the ABHD family; and Awi00008982 and Awi00009067 in the SCPL family were significantly expressed in seeds (Figure 3A).
By calculating the Ks for each duplicated gene pair associated with triterpenoid saponins synthesis, the major duplications of related genes in the synthesis pathway were generated in the recent WGD event (Figure 3B), suggesting that the occurrence of recent WGD event was significant for the evolution of triterpenoid saponins synthesis in buckeye.
Population structure and species divergence
For understanding the population structure and divergence of the main Aesculus species in China, resequencing of 104 representative samples, including 41 A. wilsonii, 31 A. chinensis, 14 A. chekiangensis, and 18 A. wangii was performed (Supplementary Table 1). These samples were sequenced to an average depth of 5.27× and coverage of 94.08% of the A. wilsonii genome. The resulted in a total of 355.63 Gb of clean data available for population analysis (Supplementary Table 1). We identified 60,387,800 high-quality single nucleotide polymorphisms (SNPs). Genetic relationships between these samples were inferred using a neighbor-joining (NJ) tree on the basis of the identified SNPs. The tree revealed that all samples were clustered into five main groups. One group included A. chekiangensis samples collected from Zhejiang and Jiangsu provinces, one group included A. wangii samples from Yunnan province, and one group was mainly A. wilsonii samples from Hubei, Henan (Figure 4A). The A. chinensis samples were divided into two distinct groups. The samples from Beijing were close to the A. chekiangensis samples, while those from Shaanxi had a close relationship with the A. wilsonii.
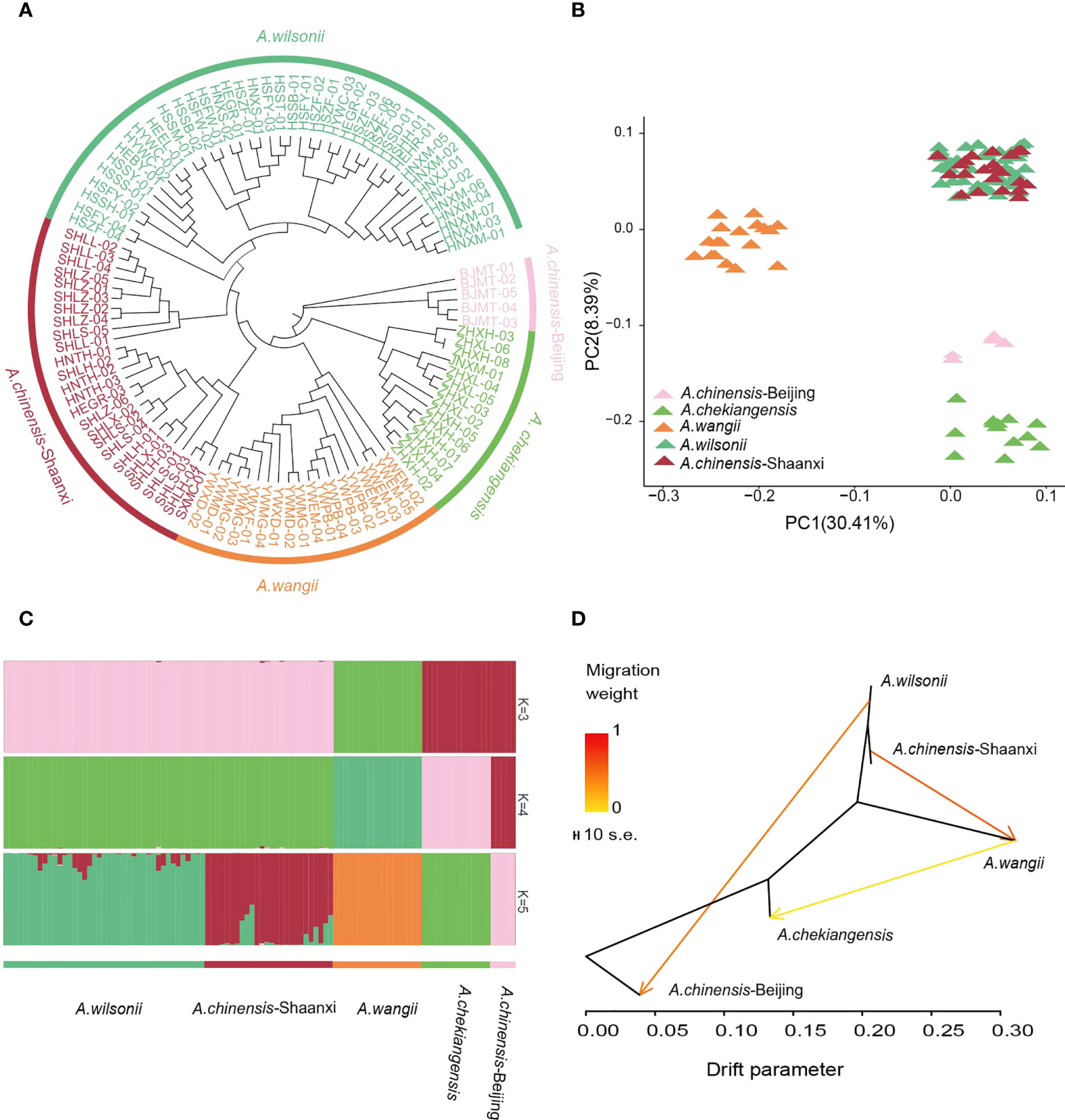
Figure 4 Population relationships and structures of buckeye. (A) Neighbor-joining phylogenetic tree of 104 buckeye accessions was constructed using SNPs. Colors represent different species. (B) PCA plots of 104 buckeye samples, with the first two principal 30.41% and 8.39%, respectively. (C) Model-based population assignment by ADMIXTURE analysis for K = 3-5. Each column represents a buckeye sample. (D) Gene-flow patterns that were detected among buckeyes groups using TreeMix. The shade of colors represented the weight of the migration events.
The ADMIXTURE analysis and principal components analysis (PCA) also confirmed this pattern (Figures 4B, C). When K = 3 in the ADMIXTURE analysis, the A. chinensis Beijing and A. chekiangensis samples, as well as the A. chinensis Shaanxi and A. wilsonii samples, were grouped into two separate groups, and both were distinct from the samples of A. wangii. When K = 4, a subdivision between the A. chinensis Beijing and A. chekiangensis samples was seen. When K = 5, the A. chinensis Shaanxi samples further diverged from the A. wilsonii samples (Figure 4C; Supplementary Figure 18). Pairwise genome-wide fixation index (FST) values between these Aesculus groups showed that genetic differentiation between A. wangii and other species groups (0.3166–0.5167) was significantly higher than other comparisons, such as those between A. chekiangensis and A. wilsonii (0.1805) and between group A. wilsonii and group A. chinensis Shaanxi (0.01300) (Supplementary Table 14). Variable levels of gene flow were observed between these specie groups (Figure 4D). Comparatively, A. wilsonii had the highest level of nucleotide diversity (π) (2.42 × 10−3) (Supplementary Table 15).
Linkage disequilibrium and demography
Linkage disequilibrium (LD, measured as r2) decreased to half of its maximum values at 368 kb in A. chinensis but at 122 kb and 94 kb in A. chekiangensis and A. wilsonii, respectively (Figure 5A). The estimated LD values of species were inversely correlated with their population nucleotide diversity (Supplementary Table 15), as expected. To investigate the demographic history of different Aesculus species in China, pairwise sequentially Markovian coalescent (PSMC) analysis was used to estimate fluctuations in the effective population size (Ne) from 20 million to 10,000 years ago. The changes in the Ne of different species over time coincided with known times of climatic events. For each species investigated, the Ne decreased in the early quaternary ice age (Ehlers and Gibbard, 2007) (∼2 Mya), and recovered during the last interglacial (∼140 kiloyears ago, kya) probably due to the rising temperatures (Petit et al., 1999). During the Last Glacial Maximum (LGM, ∼110 kya), Aesculus species populations contracted slightly, and the Ne of A. wilsonii expanded rapidly from the end of LGM (Figure 5B).
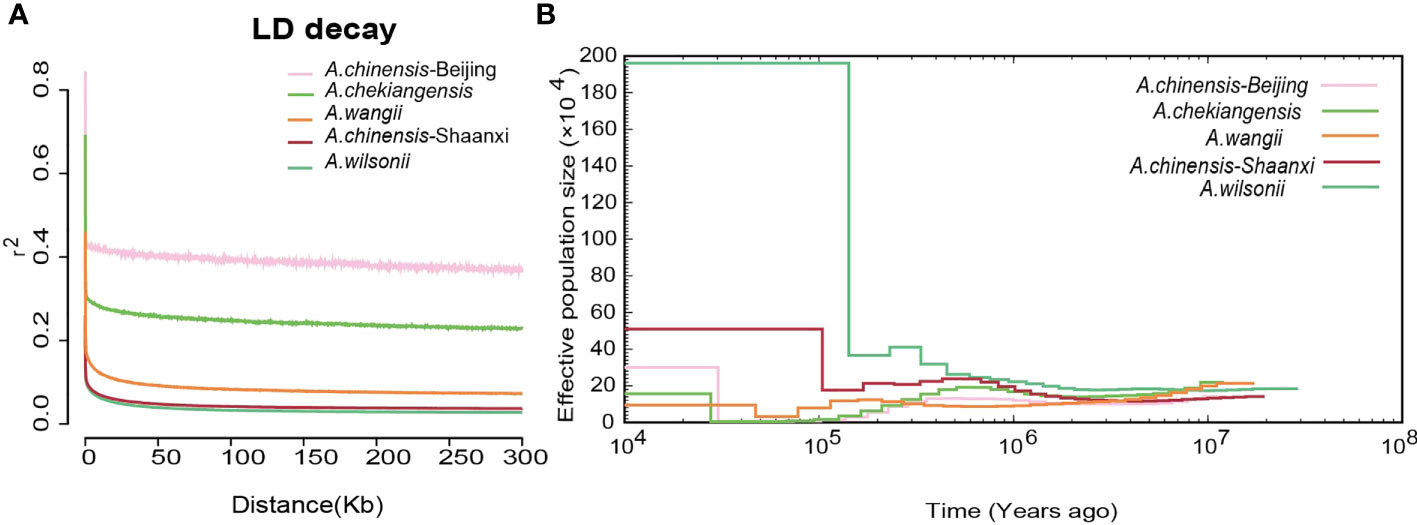
Figure 5 The Linkage disequilibrium and demography of buckeyes populations. (A) Patterns of different groups LD decay. (B) Demographic history of five buckeyes populations including A. wilsonii, A. chinensis-Shaanxi, A. wangii, A. chekiangensis, A. chinensis-Beijing.
Discussion
The Aesculus wilsonii Genome provides new resources for genetic diversity and functional studies of aesculus
Buckeye tree is known for its important medicinal and ornamental values. The seeds are rich in oil, flavonoids and saponins, and are widely used in bio-medicine and daily chemical industries. A reference genome is useful for analysis of the genetic background and secondary metabolic pathways of medicinal plants. Previous study reported that genome sizes of more than 10 species of Aesculus were previously predicted by flow cytometry (~467–623.48 Mb), and their chromosome numbers were also determined (2n = 2x = 40) (Krahulcová et al., 2017). Only the chloroplast genome of A. wilsonii (Liu et al., 2020b)and A. wangii (Zheng et al., 2017) had been reported, and no studies on the genome of buckeye tree have been reported worldwide. We present a high-quality chromosome-scale genome of A. wilsonii with the genome size of 579.01 Mb and estimated heterozygosity of 1.22%. A total of 94.72% of the assembled sequences were assigned to 20 pseudo-chromosomes with a scaffold N50 of 28.02 Mb. Compared with other species of Sapindaceae, the genome size of A. wilsonii is close to the genome size of longan (455.5 Mb, 2n=30) (Wang et al., 2022), litchi (470 Mb, 2n=30) (Hu et al., 2022), Sapindus mukorossi (432.29 Mb, 2n=28) (Xue et al., 2022), and Acer truncatum (628.84 Mb, 2n=26) (Ma et al., 2020) with a diploid genome, although the chromosome number of A. wilsonii is greater than them. On the other hand, we identified 60,387,800 high-quality SNPs by resequencing of 104 wild Aesculus samples from China, and clarified the genetic relationships between A. wilsonii and A. chinensis, and A. chekiangensis and A. wangii. These rich genomic resources for future molecular breeding and biological studies of Aesculus.
The WGD events were vital for A. wilsonii addressing environmental challenges and functional diversity
Polyploidy events are common in plants and have played an important role in plant evolution and adaptation (Wu et al., 2020). Compared with other species such longan, litchi, Sapindus mukorossi, Citrus sinensis, Acer truncatum which had only one ancient γ-WGD event, a recent specific WGD event identified in A. wilsonii (Figure 2C) was unexpected. The recent WGD event in A. wilsonii occurred at about 17.69 Mya, during the Oligocene Ice Age when the climate changed dramatically, and also when many other plant WGD events occurred (Blanc, 2004). Environmental factors such as a cold climate, climate change due to asteroid impact, and darkness are all potential drivers of plant polyploidy (Wu et al., 2020). As a result of plants responding to environmental and climatic changes, duplications of their whole genomes can rapidly increase genomic contents and genetic variation. This can provide adaptive advantages through species biased gene expression contributing to novel gene regulation networks and signal transductions (Freeling, 2009). The results of specific and expanded genes of A. wilsonii are related to light and oxygen reaction, biosynthesis of secondary metabolites, such as terpenoids, flavonoids, plant pathogen interaction, and DNA repair genes, further providing support for this conclusion. The survival probability of A. wilsonii under environmental changes may therefore benefit from the WGD event.
WGDs also contribute to the innovation of species-specific traits such as specific metabolites (Liu et al., 2021). Genomic studies of triptolide (Tu et al., 2020) and lavender (Li et al., 2021) showed that genes related to the terpenoid biosynthesis pathway were also duplicated during the WGD events. The duplication of some gene families caused by WGDs led to the diversity of terpenoids. In particular, the expansion of TPS, CYP450, and BAHD families greatly enriched the variety of terpenoids. Aescins are the only pentacyclic triterpenoid saponins existing in Aesculus. We found that the gene families related to the biosynthetic pathway of terpenoid metabolism in the genome of A. wilsonii were duplicated in different periods (Figure 3B). This was especially true for the gene families involved in the late stage of terpene synthesis such as CYP450, UGT, BAHD, and SCPL, greatly enriching the different species of pentacyclic triterpenoids.
Preliminary identification of genes specific to the biosynthesis of aescins in A. wilsonii genome
Aescins are the main characteristic active components of Aesculus species. Due to its important pharmacological activities such as anti-inflammatory, anti-edema, and inhibition of cancer cell proliferation, various pharmaceutical preparations containing aescins extract have been used in clinical treatment. Based on the A. wilsonii genome, the metabolic pathway of aescin biosynthesis was preliminarily analyzed. Analysis of unique genes in A. wilsonii genome showed that a significant enrichment of genes related to terpene biosynthesis metabolic pathways such as HMGR, MCT, GPPS, SQE, BAHD. Structural and activity analysis showed that the acyl groups on C-21 and C-22 in these saponins play an important role in their activity(Zhizhen Zhang et al., 2010). We identified Awi00015440, Awi00037368 genes in the unique genes, which belong to the IIIa subfamily of BAHD with acylation, and significantly expressed in seeds (Figures 3A, C). In other plants, homologous genes have been shown to have acylated functions (D'Auria, 2006).
Reclassification of buckeye in major regions of china and the bottleneck in the evolutionary of some population
Population genomics analysis revealed the clear genetic relationship of Aesculus species in China except for A. chinensis (Figure 4A). The Aesculus plants naturally occurring in Beijing and Shaanxi were traditionally identified as A. chinensis (FANG, 1981). However, our results showed that the samples of A. chinensis collected from Beijing and southern Shaanxi were closer to those of A. chekiangensis and A. wilsonii, respectively (Figures 4B, C). Given the relatively contiguous or overlapping geographical distributions of A. wilsonii (mainly in western Hubei and southwest Henan) and A. chinensis (from southern Shaanxi), these samples may belong to the A. wilsonii species. The slight morphological differences between them may be due to the influence of niche differentiation (Du et al., 2020). The close relationship between the A. chinensis Beijing and the A. chekiangensis samples is consistent with the record that A. chekiangensis is a variety of A. chinensis (FANG, 1981). Compared to A. wangii from Yunnan, the overall genetic differentiation between A. wilsonii, A. chinensis, and A. chekiangensis is weaker (Supplementary Table 14). In the Pharmacopoeia of the People’s republic of China (2020 edition), the seeds of A. wilsonii, A. chinensis, and A. chekiangensis are all used as TCM of Semen Aesculi. The close relationship between them may reflect a similar genomic basis, which in turn can improve the generation of consistent quality Semen Aesculi and thus benefit human health.
The A. wilsonii populations from the Qinling Mountains had a higher nucleotide polymorphism and faster LD decay than the other species populations (Figure 5A). This result was consistent with the demographic analysis, in which the range of A. wilsonii expanded significantly in the recent past, while the population expansions of A. chinensis, A. chekiangensis, and A. wangii were much less obvious (Figure 5B). In the process of species evolution, the genetic diversity of populations is affected by many factors such as the reproduction system, genetic drift, natural selection, gene flow, and human disturbance (Nevo, 2001; Frankham R and Brisoe, 2002). Among the four Aesculus species studied here, A. wilsonii has a relatively extensive natural distribution and its populations have been further increased by widespread artificial plantings in the Qinling mountain area. This is because its medicinal and ornamental values have long been recognized by local people. In contrast to A. wilsonii, the natural distribution of A. chinensis, A. chekiangensis, and A. wangii is restricted and limited utilization of their seeds has slowed down their population expansion. In addition, limited pollen transmission distance and large seeds can also affect population growth and range expansion (Isagi et al., 2007), and can lead to the population of A. chinensis, A. chekiangensis, and A. wangii experiences population bottlenecks during evolution.
Conclusion
In conclusion, lineage-specific WGD events and the related candidate genes involved in the biosynthesis of triterpenoid saponins were identified by assembling to the chromosomal level high-quality A. wilsonii genome. Population resequencing of four Aesculus species in China further clarified their genetic and evolutionary relationships. In the future, we will use recombinant proteins expressed in E. coli and gene editing techniques to functionally characterize the aescins biosynthetic genes. In addition, the small size of the present study population has limited genetic diversity of Aesculus. The more samples will be collected for gene sequencing and phenotype collection analysis to further investigate their relationship and identify genes of excellent quality. These studies provide insights that should be significant for the conservation and utilization of Aesculus.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://ngdc.cncb.ac.cn/, CRA007235, https://ngdc.cncb.ac.cn/, CRA007228.
Author contributions
LY, YL, SC, and ZS conceived and supervised the project; LY, ZS collected the samples; LLY and GC performed the raw data analysis; LY and LJ analyzed the gene families; LY and YL wrote and revised the paper; BW, ZH contributed substantially to the revisions. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key Research & Development Program of China (2017YFC1701000 and 2019YFC1711100).
Conflict of interest
Author LY and GC were employed by the company Wuhan Benagen Tech Solutions Company Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2022.1022169/full#supplementary-material
References
Alexander, D. H., Novembre, J., Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19 (9), 1655–1664. doi: 10.1101/gr.094052.109
Augustin, J. M., Drok, S., Shinoda, T., Sanmiya, K., Nielsen, J. K., Khakimov, B., et al. (2012). UDP-Glycosyltransferases from the UGT73C subfamily in barbarea vulgaris catalyze sapogenin 3-O-Glucosylation in saponin-mediated insect resistance. Plant Physiol. 160 (4), 1881–1895. doi: 10.1104/pp.112.202747
Barton, B., Castle, T. (1877). The British flora Medica:A history of the medicinal plants of great britton (Piccadilly, London: Chatto and Windus).
Bie, T. D., Cristianini, N., Demuth, J. P., Hahn, W. M. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22 (10), 1269–1271. doi: 10.1093/bioinformatics/btl097
Blanc, G. (2004). Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. THE Plant Cell Online 16 (7), 1667–1678. doi: 10.1105/tpc.021345
Braga, P. C., Marabini, L., Wang, Y. Y., Lattuada, N., Calò, R., Bertelli, A., et al. (2012). Characterisation of the antioxidant effects of aesculus hippocastanum l. bark extract on the basis of radical scavenging activity, the chemiluminescence of human neutrophil bursts and lipoperoxidation assay. Eur. Rev. Med. Pharmacol. Sci. 16 suppl 3 (4), 1–9.
Chen, N. (2004). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinf. 4, 10.11–10.14. doi: 10.1002/0471250953.bi0410s05.
Cheong, D. H. J., Arfuso, F., Sethi, G., Wang, L., Hui, K. M., Kumar, A. P., et al. (2018). Molecular targets and anti-cancer potential of escin. Cancer Lett. 422, 1–8. doi: 10.1016/j.canlet.2018.02.027
Colson, E., Decroo, C., Cooper-Shepherd, D., Caulier, G., Henoumont, C., Laurent, S., et al. (2019). Discrimination of regioisomeric and stereoisomeric saponins from aesculus hippocastanum seeds by ion mobility mass spectrometry. J. Am. Soc. Mass Spectrom 30 (11), 2228–2237. doi: 10.1007/s13361-019-02310-7
Commission, C.P (2020). Pharmacopoeia of the people's republic of China (Beijing: Chinese medicine publishing house).
D'Auria, J. C. (2006). Acyltransferases in plants: a good time to be BAHD. Curr. Opin. Plant Biol. 9 (3), 331–340. doi: 10.1016/j.pbi.2006.03.016
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Bank, E., Depristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Du, Z. Y., Harris, A. J., Xiang, Q. J. (2020). Phylogenomics, co-evolution of ecological niche and morphology, and historical biogeography of buckeyes, horsechestnuts, and their relatives (Hippocastaneae, sapindaceae) and the value of RAD-seq for deep evolutionary inferences back to the late Cretaceous. Mol. Phylogenet Evol. 145, 106726. doi: 10.1016/j.ympev.2019.106726
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 5), 1792–1797. doi: 10.1093/nar/gkh340
Ehlers, J., Gibbard, P. L. (2007). The extent and chronology of Cenozoic global glaciation. Quaternary Int. 164-165, 6–20. doi: 10.1016/j.quaint.2006.10.008
FANG, W.-P. (1981). “Hippocastanaceae,” in Flora reipublicae popularis sinicae (Beijing: Science Press), 274–289.
Finn, R. D., Tate, J., Mistry, J., Coggill, P. C., Sammut, S. J., Hotz, H. R., et al. (2008). The pfam protein families database. Nucleic Acids Res. 32 (1), D138. doi: 10.1093/nar/gkm960.
Flynn, J. M., Hubley, R., Rosen, J., Clark, A. G., Smit, A. F. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117 (17), 201921046. doi: 10.1073/pnas.1921046117
Frankham R, B. J. D., Brisoe, D. A. (2002). Introduction to con-servation genetics (Cambridge: Cambridge UniversityPress).
Fraser, C. M., Rider, L. W., Chapple, C. (2005). An expression and bioinformatics analysis of the arabidopsis serine carboxypeptidase-like gene family. Plant Physiol. 138 (2), 1136–1148. doi: 10.1104/pp.104.057950
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453. doi: 10.1146/annurev.arplant.043008.092122
Gallelli, L. (2019). Escin: a review of its anti-edematous, anti-inflammatory, and venotonic properties. Drug Des. Devel Ther. 13, 3425–3437. doi: 10.2147/DDDT.S207720
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Amit, I. (2013). Trinity: reconstructing a full-length transcriptome without a genome from RNA-seq data. Nat. Biotechnol. 29, 644. doi: 10.1038/nbt.1883
Gruza, M. M., Jatczak, K., Zagrodzki, B., Laszcz, M., Koziak, K., Malinska, M., et al. (2013). Preparation, purification and regioselective functionalization of protoescigenin–the main aglycone of escin complex. Molecules 18 (4), 4389–4402. doi: 10.3390/molecules18044389
Haralampidis, K., Trojanowska, M., Osbourn, A. E. (2002). Biosynthesis of triterpenoid saponins in plants. Adv. Biochem. Eng. Biotechnol. 75, 31. doi: 10.1007/3-540-44604-4_2
Hardin, J. W. (1957a). A revision of the American hippocastanaceae. Brittonia 9, 145–171. doi: 10.2307/2804781
Hardin, J. W. (1957b). A revision of the American hippocastanaceae-II. Brittonia 9, 173–195. doi: 10.2307/2804722
Hardin, J. W. (1957c). Studies in the hippocastanaceae, IV. hybridization in aesculus. Rhodora 59, 185–203.
Harris, A. J., Thomas, X. D. T. (2009). Phylogeny, origin, and biogeographic history of aesculus l. (Sapindales) - an update from combined analysis of DNA sequences, morphology, and fossils. Taxon 58 (1), 108–126. doi: 10.1002/tax.581012
Hoff, K. J., Simone, L., Alexandre, L., Mark, B., Mario, S. (2016). BRAKER1: Unsupervised RNA-Seq-Based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics 32 (5), 767–769. doi: 10.1093/bioinformatics/btv661
Hu, G., Feng, J., Xiang, X., Wang, J., Salojarvi, J., Liu, C., et al. (2022). Two divergent haplotypes from a highly heterozygous lychee genome suggest independent domestication events for early and late-maturing cultivars. Nat. Genet. 54 (1), 73–83. doi: 10.1038/s41588-021-00971-3
Idris, S., Mishra, A., Khushtar, M. (2020). Phytochemical, ethanomedicinal and pharmacological applications of escin from aesculus hippocastanum l. towards future medicine. J. Basic Clin. Physiol. Pharmacol. 31 (5), 20190115. doi: 10.1515/jbcpp-2019-0115
Ioanna, K., Nawrocki, E. P., Argasinska, J., Quinones-Olvera, N., Finn, R. D., Bateman, A., et al. (2018). Non-coding RNA analysis using theRfam database. Curr. Protoc. Bioinf. 62, e51. doi: 10.1002/cpbi.51
Isagi, Y., Saito, D., Kawaguchi, H., Tateno, R., Watanabe, S. (2007). Effective pollen dispersal is enhanced by the genetic structure of an aesculus turbinata population. J. Ecol. 95 (5), 983–990. doi: 10.1111/j.1365-2745.2007.01272.x
Johnson, V. E., Tuominen, L. K., Chung-Jui, T. (2011). Differential phylogenetic expansions in BAHD acyltransferases across five angiosperm taxa and evidence of divergent expression among populus paralogues. BMC Genomics 12 (1), 236. doi: 10.1186/1471-2164-12-236
Jozwiak, A., Sonawane, P. D., Panda, S., Garagounis, C., Papadopoulou, K. K., Abebie, B., et al. (2020). Plant terpenoid metabolism co-opts a component of the cell wall biosynthesis machinery. Nat. Chem. Biol. 16 (7), 740–748. doi: 10.1038/s41589-020-0541-x
Kevin, L., Randal, L. C., Tandy, W., Rongling, W. (2011). RAxML and FastTree: Comparing two methods for Large-scale maximum likelihood phylogeny estimation. PloS One 6 (11), e27731. doi: 10.1371/journal.pone.0027731.
Kondrashov, N., Pusic, A., Stumpf, C. R., Shimizu, K., Hsieh, A., Xue, S., et al. (2011). Ribosome-mediated specificity in hox mRNA translation and vertebrate tissue patterning. Cell 154, 383–387. doi: 10.1016/j.cell.2011.03.028
Krahulcová, A., Trávníček, P., Krahulec, F., Rejmánek, M. (2017). Small genomes and large seeds: chromosome numbers, genome size and seed mass in diploid aesculus species (Sapindaceae). Ann. Bot. 119, 957–964. doi:10.1093/aob/mcw261
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Li, S. (1982). Bencao gangmu-compendium of materia medica. Beijing: People's Medical Publishing House.
Li, H., Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). Genome project data processing s: The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, L., Stoeckert, C., Roos, D. S. (2003). OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13 (9), 2178–2189. doi: 10.1101/gr.1224503
Liu, B., Shi, Y., Yuan, J., Hu, X., Zhang, H., Li, N., et al. (2013). Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Quantitative Biol. 35 (s 1–3), 62–67. doi: 10.48550/arXiv.1308.2012
Liu, Z., Suarez Duran, H. G., Harnvanichvech, Y., Stephenson, M. J., Schranz, M. E., Nelson, D., et al. (2020a). Drivers of metabolic diversification: how dynamic genomic neighbourhoods generate new biosynthetic pathways in the brassicaceae. New Phytol. 227 (4), 1109–1123. doi: 10.1111/nph.16338
Liu, Y., Wang, B., Shu, S., Li, Z., Song, C., Liu, D., et al. (2021). Analysis of the coptis chinensis genome reveals the diversification of protoberberine-type alkaloids. Nat. Commun. 12 (1), 3276. doi: 10.1038/s41467-021-23611-0
Liu, Z., Zhang, J., Zhou, Y., Liu, Y., Hu, Z., Zheng, G., et al. (2020b). The complete chloroplast genome of aesculus chinensis var. wilsonii. Mitochondrial DNA B Resour 5 (3), 2547–2549. doi: 10.1080/23802359.2020.1780972
Li, J., Wang, Y., Dong, Y., Zhang, W., Wang, D., Bai, H., et al. (2021). The chromosome-based lavender genome provides new insights into lamiaceae evolution and terpenoid biosynthesis. Hortic. Res. 8 (1), 53. doi: 10.1038/s41438-021-00490-6
Lowe, T. M., Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25 (5), 955–964. doi: 10.1093/nar/25.5.955
Ma, Q., Sun, T., Li, S., Wen, J., Zhu, L., Yin, T., et al. (2020). The acer truncatum genome provides insights into nervonic acid biosynthesis. Plant J. 104 (3), 662–678. doi: 10.1111/tpj.14954
Minoru, K., Susumu, G., Yoko, S., Masayuki, K., Miho, F., Mao, T. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Nawrocki, E. P., Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935. doi: 10.1093/bioinformatics/btt509
Nelson, D., Werck-Reichhart, D. (2011). A P450-centric view of plant evolution. Plant J. 66 (1), 194–211. doi: 10.1111/j.1365-313X.2011.04529.x
Nevo, E. (2001). Evolution of genome–phenome diversity under environmental stress. Proc. Natl. Acad. Sci. United States America 98 (11), 6233–6240. doi: 10.1073/pnas.101109298
Nutzmann, H. W., Huang, A., Osbourn, A. (2016). Plant metabolic clusters - from genetics to genomics. New Phytol. 211 (3), 771–789. doi: 10.1111/nph.13981
Nutzmann, H. W., Scazzocchio, C., Osbourn, A. (2018). Metabolic gene clusters in eukaryotes. Annu. Rev. Genet. 52, 159–183. doi: 10.1146/annurev-genet-120417-031237
Petit, J. R., Jouzel, J., Raynaud, D., Barkov, N. I., Barnola, J. M., Basile, I., et al. (1999). Climate and atmospheric history of the past 420,000 years from the vostok ice core, Antarctica. Nature 399 (6735), 429–436. doi: 10.1038/20859
Pickrell, J. K., Pritchard, J. K., Tang, H. (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PloS Genet. 8 (11), e1002967. doi: 10.1371/journal.pgen.1002967
Porebski, S., Bailey, L. G., Baum, B. R. (1997). Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Rep. 15 (1), 8–15. doi: 10.1007/BF02772108
Pu, X., Dong, X., Li, Q., Chen, Z., Liu, L. (2021). An update on the function and regulation of methylerythritol phosphate and mevalonate pathways and their evolutionary dynamics. J. Integr. Plant Biol. 63 (7), 1211–1226. doi: 10.1111/jipb.13076
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M., Bender, D., et al. (2007). PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi: 10.1086/519795
Schiffels, S., Durbin, R. (2014). Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 46, 919–925. doi: 10.1038/ng.3015
Schmidt, H.-W. M., Vogel, A., Denton, A. K., Istace, B., Wormit, A., Geest, V. D. H., et al. (2017). De novo assembly of a new solanum pennellii accession using nanopore sequencing. Plant Cell 10, 2336–2348. doi: 10.1105/tpc.17.00521
Simão, F. A., Waterhouse, R. M., Panagiotis, I., Kriventseva, E. V., Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31 (19), 3210–3212. doi: 10.1093/bioinformatics/btv351
Sudhir, K., Glen, S., Michael, S., Blair, H. S. (2017). TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34 (7), 1812–1819. doi: 10.1093/molbev/msx116
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., Paterson, A. H. (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Thimmappa, R., Geisler, K., Louveau, T., O'Maille, P., Osbourn, A. (2014). Triterpene biosynthesis in plants. Annu. Rev. Plant Biol. 65, 225–257. doi: 10.1146/annurev-arplant-050312-120229
Tu, L., Su, P., Zhang, Z., Gao, L., Wang, J., Hu, T., et al. (2020). Genome of tripterygium wilfordii and identification of cytochrome P450 involved in triptolide biosynthesis. Nat. Commun. 11 (1), 971. doi: 10.1038/s41467-020-14776-1
Vaser, R., Sovic, I., Nagarajan, N., Sikic, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27 (5),737–746. doi: 10.1101/gr.214270.116
Vos, P. G., Paolo, M. J., Voorrips, R. E., Visser, R. G., van Eck, H. J., van Eeuwijk, F. A. (2017). Evaluation of LD decay and various LD-decay estimators in simulated and SNP-array data of tetraploid potato. Theor. Appl. Genet. 130, 123–135. doi: 10.1007/s00122-016-2798-8
Walker, B. J., Abeel, T., Shea, T., Priest, M., Earl, A. M. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One 9 (11), e112963. doi: 10.1371/journal.pone.0112963
Wang, J., Li, J., Li, Z., Liu, B., Zhang, L., Guo, D., et al. (2022). Genomic insights into longan evolution from a chromosome-level genome assembly and population genomics of longan accessions. Hortic. Res. 9, uhac021. doi: 10.1093/hr/uhac021
Wu, S., Han, B., Jiao, Y. (2020). Genetic contribution of paleopolyploidy to adaptive evolution in angiosperms. Mol. Plant 13 (1), 59–71. doi: 10.1016/j.molp.2019.10.012
Xiang, Q.-Y., Crawford, D. J., Wolfe, A. D., Tang, Y.-C., DePamphlils, C. W. (1998). Origin and biogeography of aesculus l. (Hippocastanaceae): A molecular phylogenetic perspective. Evolution 52, 988–997. doi: 10.1111/j.1558-5646.1998.tb01828.x
Xia, N., Turland, N. (2005). A new combination in Chinese aesculus (Hippocastanaceae). Novon 15, 488–489.
Xue, T., Chen, D., Zhang, T., Chen, Y., Fan, H., Huang, Y., et al. (2022). Chromosome-scale assembly and population diversity analyses provide insights into the evolution of sapindus mukorossi. Hortic. Res. 9, uhac012. doi: 10.1093/hr/uhac012
Xu, R., Fazio, G. C., Matsuda, S. P. (2004). On the origins of triterpenoid skeletal diversity. Phytochemistry 65 (3), 261–291. doi: 10.1016/j.phytochem.2003.11.014
Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24 (8), 1586–1591. doi: 10.1093/molbev/msm088
Yang, J., Lee, S. H., Goddard, M. E., Visscher, P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88 (1), 76–82. doi: 10.1016/j.ajhg.2010.11.011
Yu, G. (2020). Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinf. 69 (1), e96. doi: 10.1002/cpbi.96
Zdobnov, E. M., Rolf, A. (2001). InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 9), 847–848. doi: 10.1093/bioinformatics/17.9.847
Zhang, Z., Li, S., Lian, X. (2010). An overview of genus aesculus l.: Ethnobotany, phytochemistry, and pharmacological activities. Pharm. Crops 1 (1), 24–51. doi: 10.2174/2210290601001010024
Zhang, F. S., Zhang, X., Wang, Q. Y., Pu, Y. J., Ma, C. G. (2020). Cloning, yeast expression, and characterization of a β-amyrin c-28 oxidase (CYP716A249) involved in triterpenoid biosynthesis in polygala tenuifolia. Biol. Pharm. Bull. 43 (12), 1839–1846. doi: 10.1248/bpb.b20-00343
Zhang, X., Zhang, S., Zhao, Q., Ming, R., Tang, H. (2019). Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-c data. Nat. Plants 5 (5), 833–845. doi: 10.1038/s41477-019-0487-8
Keywords: Aesculus wilsonii, chromosome-level genome, whole-genome duplication, terpenoid biosynthesis, aescins, phylogenetic relationship
Citation: Ye L, Yang L, Wang B, Chen G, Jiang L, Hu Z, Shi Z, Liu Y and Chen S (2022) The Chromosome-level genome of Aesculus wilsonii provides new insights into terpenoid biosynthesis and Aesculus evolution. Front. Plant Sci. 13:1022169. doi: 10.3389/fpls.2022.1022169
Received: 18 August 2022; Accepted: 12 September 2022;
Published: 18 October 2022.
Edited by:
Shaojun Dai, Shanghai Normal University, ChinaReviewed by:
Mei Yang, Wuhan Botanical Garden (CAS), ChinaMuhammad Amjad Nawaz, Far Eastern Federal University, Russia
Copyright © 2022 Ye, Yang, Wang, Chen, Jiang, Hu, Shi, Liu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaohua Shi, zhshi78@hbtcm.edu.cn; Shilin Chen, slchen@icmm.ac.cn; Yifei Liu, liuyifei@hbtcm.edu.cn
†These authors have contributed equally to this work