
Abstract
On the island of Hispaniola, five endemic species of Magnolia occur, all of which are threatened with extinction. Little is known about their distribution and genetic health, hampering targeted conservation actions. The objective of this study is to assess the potential distribution and the genetic health of the Magnolias of Hispaniola, to inform concrete guidelines for effective conservation management. Using species distribution modelling (SDM), we predict habitat suitability for the Magnolias of Hispaniola by analysing 21 variables, describing climate and landscape features, on 635 occurrences. We genotyped 417 individuals using 16 microsatellite markers, to test for genetic structure and degree of inbreeding. The SDM and genetic data confirm the recognition of the four studied Magnolia species. The known individuals of the three Dominican Magnolias are structured into five populations which show ample genetic diversity and little inbreeding overall. For conservation management, we propose to focus on exploration using the SDM results, and protection and reinforcement using the genetic and occurrence data. The genetic results guide prioritization of species and populations. The SDM results guide spatial prioritization. Installing and/or protecting habitat corridors between populations, starting with the two species with the lowest genetic diversity and relatively nearby populations, is recommended as a durable conservation strategy. Meanwhile, reinforcement efforts can be undertaken to artificially increase gene flow for which we appoint sink and source population pairs, using the genetic data.
Similar content being viewed by others

Introduction
Tropical montane cloud forests (henceforth TMCFs) comprise 0.26% of the Earth’s surface, yet they are home to a disproportionately high number of species (Bubb et al. 2004). This habitat is characterized by heavy rainfall, high species endemism and low resilience to disturbance (Hamilton et al. 1995). Despite their importance of harbouring high numbers of (endemic) biodiversity, TMCFs face similar threats as many other tropical forest habitats (Bubb et al. 2004). While progress on the sustainable conservation of TMCFS is being made (López-Barrera et al. 2017), many challenges still need to be overcome (Hamilton et al. 1995). For example, Wilson and Rhemtulla (2018) showed that larger and more intact cloud forests contained more tree species, but small remnant forest patches still contribute significantly to biodiversity. They concluded that the small remnant forest patches are essential building-blocks for reserve design aimed at conserving landscape-level tree diversity. Yet, rising anthropogenic pressures are altering forest species compositions and ecosystem functioning (Malhi et al. 2014). To be effective, management mitigation and restoration actions need, amongst others, information on the current and potential distribution of high-quality areas, as well as on the current state of remnant populations, information which is often unavailable (Toledo-Aceves et al. 2021).
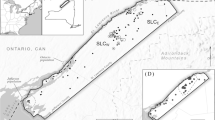
One area that exemplifies the situation of TMCFs and the challenge to their conservation is the island of Hispaniola, the second largest Caribbean Island. It is divided in two countries, Haiti and the Dominican Republic (henceforth DR). Haiti is one of the most deforested countries on earth, with less than 1% of its original primary forests remaining (Hedges et al. 2018), yet the DR suffers likewise from forest loss and degradation (Sangermano et al. 2015a). Nonetheless, Hispaniola remains a biodiversity hotspot due to its high species endemism, housing for example about 6000 endemic plant species (Maunder et al. 2008; Cano-Ortiz et al. 2016, 2017). Five of these are endemic Magnolia species: M. domingensis, M. ekmanii, M. emarginata, M. hamorii and M. pallescens (Castillo 2016), which are all threatened to some degree (Rivers et al. 2016) and consisting of only a few (known) populations each (Fig. 1). The Magnolias of Hispaniola are at great risk of extinction, with one of the most prevalent factors being habitat loss due to timber exploitation and land conversion (Rivers et al. 2016; Castillo et al. 2018; Veltjen et al. 2019). As these species reside in TMCFs, their survival is intricately linked to the preservation of this habitat. Because Magnolia is an eye-catching genus due to its beautiful flowers, aromatic leaves and ornamental use (Fig. 2), it serves as an umbrella species for conservation (Roberge and Angelstam 2004), safeguarding both other species co-occurring in the TMCFs, and the habitat itself.
To construct effective conservation strategies, several sources of information are essential: identification of the most important threats, a clear delineation of species identity, sufficient knowledge on the species’ habitat requirements and their current and potential geographical distribution, and a quantification of the genetic diversity of remnant populations (Kramer and Havens 2009; IUCN 2017). Ideally, all this information is known and allows one to confidently and correctly define ‘management units’, i.e., groups of individuals that are clustered together for conservation management purposes to contain the genetic diversity necessary to ensure evolvability in the light of changing environments and local adaptation (Weckworth et al. 2018). While some data on the geographical distribution of the Magnolias of Hispaniola are available (Castillo et al. 2018; Veltjen et al. 2019), their potential and actual distribution remain unclear. Similar doubts exist regarding the genetic diversity and structure of the currently known populations. Veltjen et al. (2019) assessed the population structure and genetic health for all extant Caribbean Magnolia species, including four out of five Magnolia species from Hispaniola (Magnolia emarginata was not assessed as individuals of this Haitian species have only been rediscovered by Eladio M. Fernández in June 2022 after being lost to science for 97 years (Nowshin 2022)). However, the Veltjen et al. (2019) study used only two localities per species, with 20 individuals genotyped for each locality, whilst much more localities and individuals are currently known. This allowed to deduct a general genetic pattern for the Caribbean Magnolias (i.e., little inbreeding, yet high genetic structuring), but species-specific recommendations could not be presented as the data were too limited. To provide clarity on the species delimitation, distribution and genetic diversity of the known remnant populations of the Magnolias of Hispaniola, we integrate in this study species distribution modelling and conservation genetics techniques.
Species distribution modelling (SDM) is widely used to address questions in conservation biology, ecology, and evolution (Guisan and Thuiller 2005; Guillera-Arroita et al. 2015). It allows to predict the species’ (potential) distributions and the habitat suitability, based on species occurrence data and environmental information. Conservation genetics can resolve fragmented population structure and identify species or populations at risk as a result of reduced genetic diversity (Frankham et al. 2015). Compared to Veltjen et al. (2019), we significantly expand here the sampling of individuals, number of localities and SSR markers, to more comprehensively and accurately quantify the genetic diversity of M. domingensis, M. hamorii and M. pallescens (Medina et al. 2006; Wang et al. 2021). By integrating the results of the SDM and the comprehensive conservation genetic analyses, we formulate concrete conservation actions that maximize the genetic diversity in the long term. In doing so, we strive to maintain healthy populations with enough evolvability to make them flexible and resistant to future disturbances.
We addressed the following objectives regarding the distribution and genetic health of the Magnolias of Hispaniola. Using SDM, we aimed to (1) predict suitable Magnolia habitat for conservation efforts and to guide explorations for new populations. Furthermore, using conservation genetics, we aimed to (2) assess the population structure to infer conservation units, and (3) quantify genetic diversity, enabling us to make statements about the genetic health of the populations. Finally, we integrated results of both disciplines to (4) formulate concrete conservation management actions.
Materials and methods
Species distribution modelling
Data preparation: firstly, occurrence data needed to be rarefied over a predefined rarefying distance to minimize spatial autocorrelation. Available occurrence data are often not gathered using systematic surveying methods, but include opportunistic data and typically do not represent a random sampling across the species’ range (Phillips et al. 2009). Occurrence data are thus often biased in terms of sampling effort. Without rarefaction, this bias will cause predictions to overestimate habitat suitability in environments similar to the more intensely sampled areas (Elith and Leathwick 2009). Secondly, the SDM algorithms needed information about the absence of species in the region, which typically are only rarely available (Zurell 2020). To tackle this lack of reliable absence data, “pseudo-absences” (also referred to as “background data”) are generated from a predefined background area (Barbet-Massin et al. 2012). The selection of a proper prevalence ratio (i.e., the number of pseudo-absences relative to the number of presences) is the subject of ongoing debate, but mainly depends on the SDM algorithms applied (Liu et al. 2019). Therefore, we tested different pseudo-absence selection strategies in accordance with Lobo et al. (2010).
Model fitting and evaluation: as algorithms have different underlying assumptions and extrapolate to unsampled environments in a different manner, it has been proposed to combine predictions across multiple modelling methods. This “ensemble modelling” enhances predictive performance and reduces uncertainty (e.g. Hao et al. 2020) in model forecasts. We built and evaluated the performance of the modelling algorithms by cross-validating the data. This model fitting and evaluation strategy divides the data in two subsets: a training set, which trains the model, and an independent test set, which tests its predictive accuracy. Finally, an ensemble of SDM models that agrees with a predefined minimum predictive accuracy was used to obtain final predictions of Magnolia habitat suitability and potential geographic distribution across the study area.
Despite our focus on Magnolias from the Dominican Republic, SDM was executed on Hispaniola as a whole (i.e., including Haiti) as delineating study areas based on ecologically meaningless criteria such as country borders risk resulting in spurious predictions of habitat suitability (Bystriakova et al. 2012).
Occurrence data and predictor variables
Occurrence data of four of the five species from Hispaniola were obtained from Veltjen (2020), Castillo et al. (2018), herbarium records (Online Resource 1) and pers. comm. of Joel Timyan (Société Audubon Haiti). Occurrence data were retained if they had a spatial resolution of ≤ 1” or ~ 0.0083333°. No occurrence data of M. emarginata were considered because there were no recent nor sufficiently precise locality data at the time of our analyses. Similarly, no occurrence data of M. domingensis from Haiti were included. This led to a presence-only dataset of 635 occurrences. This dataset was subsequently rarefied with a rarefying distance of 1 km, reducing it to 30 independent occurrences. Because a minimum of five independent occurrences per species are necessary after rarefying (Broennimann et al. 2012), we opted for an explicit “genus-level”-approach (Phillips et al. 2017; Stas et al. 2020) and performed SDM on all remaining independent occurrences.

Map visualizing four Magnolia species from Hispaniola. White triangles are mountain ranges. Base layer depicts elevation as meters above sea level (m.a.s.l.)
Predictor variables were obtained from ENVIREM (Title and Bemmels 2018), CHELSA (Karger et al. 2017) and SEDAC (Venter et al. 2016, 2018). The included environmental variables describe temperature, precipitation, aridity, evapotranspiration, and topography, and one variable describing cumulative human pressures on the environment. To identify strongly correlated variables, we used the “raster.cor.matrix” and “raster.cor.plot” function in the R package ENMTools (Warren et al. 2010). The Pearson correlation coefficient “r” was used to determine which variables were most strongly correlated (|r| > 0.7). Of the correlated variables, we selected the most ecophysiologically meaningful ones in accordance with Mod et al. (2016) (see Online Resource 2). All variables were obtained with a spatial resolution of 1 km2 (~ 0.0083333°) or less.

Leaf and flower morphology of three of the five Magnolia species from Hispaniola. Classification follows Figlar and Nooteboom (2004). a, b. Magnolia domingensis. (a) Flower in male phase. (b) Villose leaves. c, d. Magnolia hamorii. (c) Closed flower bud. (d) Leaves and evidence of conduplicate leaf prefoliation. e, f. Magnolia pallescens. (e) Immature flower with underdeveloped reproductive structures. (f) Elliptic leaf with abaxial sericeous pubescence. Photographs: 2a & 2b– Ramón Elías Castillo Torres; 2b, 2d, 2e, 2f – Emily Veltjen
Model fitting and evaluation
The different pseudo-absence selection strategies resulted in 15 ensemble models predicting the suitability of Hispaniola for the focal Magnolia species. Each ensemble model consisted of eight different modelling algorithms, as implemented in the ensemble modelling framework of the package “sdm” (Naimi and Araújo 2016) in R (R Core Team 2020): two regression-based models (Generalised Linear Model (GLM) & Multivariate Adaptive Regression Splines (MARS)), five machine-learning models (Classification and Regression Trees (CART), Boosted Regression Trees (GBM), Maximum Entropy (MaxEnt), Random Forest (RF) & Support Vector Machine (SVM)) and one likelihood-based model (Maxlike). Each SDM algorithm ran with default settings (Naimi and Araújo 2016). Pseudo-absences were sampled at random in background areas defined as either concentric zones around the presence data (using four different radii (i.e., 10 km, 25 km, 50 km & 75 km)) or across the whole of Hispaniola (i.e., no radius restriction) sensu VanDerWal et al. (2009). Pseudo-absences were not selected from pixels containing Magnolia presences. Pixels correspond with the spatial resolution of the predictor variables (i.e., 1 km2). Three prevalence ratios were used to sample pseudo-absences: neutral prevalence (1:1), twice the number of presences (1:2) and five times as many (1:5). Prevalence ratios were kept low to account for the low number of (independent) Magnolia occurrences, as proposed by Liu et al. (2019). This led to a total of 15 (5 × 3) predictions of Magnolia habitat suitability (on a scale of 0 to 1), which were combined into ensemble predictions. Each individual model was calibrated using a 10-fold cross-validation with 80 − 20% random split of the presence data to serve as training data for each replicate. Model evaluation is given by the True Skill Statistic (TSS), since it is considered the most optimal measure for the performance of predictive models (Allouche et al. 2006). TSS values range from − 1 to + 1, with values of zero or less indicating a performance that is no better than random, while a value of + 1 suggests a perfect model capacity to discriminate between suitable and unsuitable areas. Interpretation of the TSS values follows Allouche et al. (2006). For each of the 15 ensemble models, only “good performing” algorithms, characterized by a TSS-value of > 0.7, were kept and ensemble habitat suitabilities were obtained using simple averaging of the good performing algorithm outcomes.
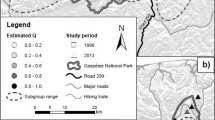
To obtain a single consensus prediction of likely Magnolia occurrence across Hispaniola, three different methods were used to summarize the 15 ensemble model outcomes above. First, the 15 Magnolia habitat suitability predictions were transformed from continuous suitabilities (0 to 1) to discrete predicted presences versus absences, using the habitat suitability threshold maximizing model TSS as cut-off and setting the suitability for pixels below this threshold to zero. Three final distribution maps were created. One obtained through “vote counting” pixels in every presence-absence prediction, thus creating a gradient from 0 (area was never suitable) to 15 (area was always indicated as suitable). A second by employing a weighted averaging procedure, combining the 15 habitat suitabilities into a single habitat suitability map, weighing individual models by their TSS values (better models contribute more strongly to the final prediction). A third by performing a similar standard “unweighted” averaging. These latter two methods resulted in a continuous prediction of habitat suitability between 0 and 1. An additional ensemble distribution map, specifically for the DR, was established from the “vote counting” distribution map of Hispaniola. Here, we added an additional layer of forest cover from the year 2000, obtained from Sangermano et al. (2015b).

Ensemble-based distribution map for the Magnolia species from Hispaniola and the Dominican Republic using weighted averaging. Habitat suitability ranges from zero to one, representing the weighted mean TSS-value for each pixel

Ensemble-based distribution map for the Dominican Republic using “vote counting”. Habitat suitability ranges from zero to fifteen, representing the number of ensemble analyses that labelled a pixel as suitable (i.e., TSS-value > 0.7). Forest 2000 layer is derived from Sangermano et al. (2015b)
Conservation genetics
Plant material and DNA extraction
Individuals of Magnolia domingensis, M. hamorii and M. pallescens in the DR were sampled in 2015 and 2021 by haphazard sampling (Ward and Jasieniuk 2009; Veltjen et al. 2019). In total, 417 individuals were genotyped, sampled at 12 distinct localities. For each locality sampled in 2015, one herbarium specimen serves as a morphological voucher for its species identification. Sample information and a map of the sampling locations are given in Table 1; Fig. 1, respectively.
DNA isolation was performed on dried leaf tissue according to a modified cetyltrimethylammonium bromide (CTAB) extraction protocol (Doyle and Doyle 1987), with MagAttract Suspension G solution (Qiagen, Germantown, USA) mediated cleaning (Larridon et al. 2015). A Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, Massachusetts, USA) and Nanodrop 2000 Spectrophotometer (Thermo Fisher Scientific) were used to assess DNA quantity and quality, respectively.
Microsatellite testing and genotypification
Twenty-five microsatellite or Simple Sequence Repeat (SSR) markers from Veltjen et al. (2019) were selected, based on the SSR marker being polymorphic in at least one of the three Magnolia species from the DR. From the localities sampled in 2015 (i.e., BAR, ROD, CAC, COR, CAS, SAL and MON in Table 1), a selection of 24 individuals was used to re-evaluate the performance of these microsatellite markers (Online Resource 3). The 24 individuals were chosen to include as much variation as possible on a spatial scale and to have 260/230 and 260/280 optical density (OD) ratios approaching 2. Performance testing was done using simplex (i.e., one microsatellite marker per PCR) three-primer PCRs (Vartia et al. 2014). A reaction contained 2 × QIA Multiplex PCR Master Mix (Qiagen), 5ng/µl DNA, 0.025 µM for each forward primer, 0.1 µM for each reverse primer and 0.1 µM for each specified dye. The selected primer pairs were labelled with a fluorescent dye FAM, NED, PET and VIC, which were linked to their respective universal tail T3, Hill, Neo and M13. PCRs ran on a volume of 5 µl under the following conditions: an initial activation step of 15 min at 95 °C, followed by 35 cycles of 30 s at 94 °C, 90 s at 57 °C and 90 s at 72 °C; and a final extension for 10 min at 72 °C. An ABI 3730XL fragment analyser (Thermo Fisher Scientific) with a GeneScanTM 500 LIZ™ ladder (Thermo Fisher Scientific) was used to quantify the PCR products. The results of these simplex tests were analysed in Geneious v.8.1.9 (http://www.geneious.com, Kearse et al. 2012) using the microsatellite plugin. An SSR marker was included in the subsequent multiplex design and final genotyping if it could be unambiguously scored for all tested species and populations, and if it was polymorphic. Multiplex pools were designed using Multiplex Manager (Holleley and Geerts 2009). PCR conditions and peak calling occurred cf. the simplex testing above. In total, four multiplex pools allowed unambiguous genotypification of the test-individuals, whereafter we genotyped the full dataset of 417 individuals. In total we evaluated 25 SSR markers of which 17 were considered qualitative and unambiguous upon constructing the final dataset.
To account for human and technical flaws such as genotyping errors and null alleles, we ran MICROCHECKER v.2.2.3 (Van Oosterhout et al. 2004) and ML-NullFreq (Kalinowski and Taper 2006). Null alleles are alleles that do not produce a functional end-product or have a mutation in the primer region which may inhibit PCR amplification (De Meeûs 2018; Frankham et al. 2015). MICROCHECKER and ML-NullFreq ran for 1000 and 100,000 repetitions, respectively. No markers were removed. MICROCHECKER labelled five markers with potential null alleles: MA40_045, MA42_001, MA42_059, MA42_126 and MA42_472. However, they were only highlighted in locality MON. ML-NullFreq indicated high values (> 0.1) for MA42_001 and MA42_059 in locality MON and for MA42_203, MA42_241 and MA42_397 in locality TON. TON is a locality with a small sample size; hence, we did not consider these as a true indication for null alleles. Similarly, as null alleles were only found in MON and in no other locality, no markers were removed from the subsequent downstream analyses.
To assure random sampling of the genome, we examined the dataset for the presence of linkage disequilibrium (LD) using the program GENEPOP v.4.7.5 (Raymond and Rousset 1995; Rousset 2008). GENEPOP ran with the dememorization number set to 10,000, with 1000 batches and with 50,000 iterations per batch. Following Waples (2015), both uncorrected and (sequential Bonferroni) corrected p-values were considered. GENEPOP revealed that one marker, MA40_045, showed significant LD with three markers after sequential Bonferroni correction: MA42_231 in ROD, MA40_282 in BAR and MA42_472 in ROD, SAL, and MON. Hence, MA40_045 was discarded to guarantee independent sampling of the genome with respect to other microsatellite markers, bringing the final number of markers to a total of 16 (see Online Resource 4).
Population structure and genetic diversity
To infer the number of conservation units, we assessed the population structure in three ways: STRUCTURE analyses, DAPC analyses and genetic differentiation measurements. In STRUCTURE v.2.3.4 (Pritchard et al. 2000), analyses were executed using four datasets: one with all three focal species and three datasets for each species. STRUCTURE ran under the following conditions: 100 000 Markov chain Monte Carlo (MCMC) replicates after an initial burn-in of 100 000, correlated allelic frequencies and the admixture model. Correlated allele frequencies were selected, given the little phylogenetic differentiation (Veltjen et al. 2022) and conservation genetic differentiation (Veltjen et al. 2019) between the three species. The number of groups (K) was set from one to twenty for the general dataset (all species included), which allowed all 12 sampling localities to be retrieved as a separate population and to account for the possibility of undetected substructures. K was set from one to ten for the species-specific datasets. Each value of K was run ten times for each dataset. A visualization of the results was obtained from Structure Harvester Web v.0.6.94 (Earl and vonHoldt 2012). The optimal K-value was selected based on the ΔK statistic, following Evanno et al. (2005), and the mean maximum likelihood (Mean LnK or \(\stackrel{-}{\text{L}\text{n}\text{K}}\)). Mean LnK proved valuable as ΔK cannot retrieve K = 1 as the most optimal K-value. Visualisation of the STRUCTURE barplots was done with DISTRUCT v.1.1 (Rosenberg 2004). DAPC (Discriminant Analysis of Principal Components) analyses were executed in R using the package “adegenet” (Jombart and Ahmed 2011) on the same datasets as the STRUCTURE analyses. Using the “find.cluster” function, we retained the maximum number of PCs and selected groups on the basis of the Bayesian Information Criterion (BIC). The number of Principal Component Analysis (PCA) eigenvalues was determined using 1000 cross-validation replicates. The PCA of the DAPC was set according to the lowest Mean Squared Error (MSE). All discriminant functions (i.e., DA eigenvalues) were kept. Genetic differentiation measurements comprised: pairwise FST (Weir and Cockerham 1984), GST (Nei and Chesser 1983), G’ST (Hedrick 2005) and Jost’s D (DJ; Jost 2008). The values and their confidence intervals were calculated for each population and locality using the R package diveRsity (Keenan et al. 2013). Interpretation of the FST-values follows Hartl and Clark (1997): <0.05 is considered “little”, 0.05–0.15 “moderate”, 0.15–0.25 “great” and > 0.25 “very great” genetic differentiation.
To characterize the genetic health, we quantified the genetic diversity with diversity statistics for each locality and population as defined in Table 1 and the population structure analyses, respectively. Genetic diversity was estimated by quantifying the following statistics in GenAlEx v.6.503 (Peakall and Smouse 2006, 2012): number of alleles per locus (A), number of private alleles (AP), observed heterozygosity (HO), expected heterozygosity (HE) and the number of polymorphic loci (P). Moreover, allelic richness (AR) and the inbreeding coefficient (FIS) were calculated in FSTAT v.2.9.4 (Goudet 2000). AR−X represents the allelic richness rarefied to X number of individuals. To quantify if FIS significantly differed from zero, deviations from Hardy-Weinberg proportions (HWP) were tested in GENEPOP with 2-tailed exact tests for every marker × population combination. If possible, a complete enumeration was executed (Louis and Dempster 1987). Otherwise, MCMC chains were employed with 200 batches and 50 000 iterations (Guo and Thompson 1992).
Results
Species distribution modelling
Out of 39 candidate predictor variables, 21 were retained because they were uncorrelated and considered (more) ecophysiologically meaningful (Online Resource 2). During model evaluation, no ensemble models were discarded as all 15 models had a model accuracy of TSS ≥ 0.7. Values for model performance across prevalence ratios and pseudo-absence sampling distance range from 0.800 to 0.993 (Table 2). These values exceed 0.7, and therefore indicate good predictive models (Allouche et al. 2006). Model performance for each of the 15 ensemble predictions can be found in Table 2. TSS-values correlated positively with increasing pseudo-absence sampling distance as well as higher prevalence ratios. The three final SDM maps based on vote counting, weighted, and unweighted averaging can be found in Fig. 3 and Online Resource 5. The additional ensemble distribution map for the DR can be found in Fig. 4.
Population structure and genetic diversity
For all four STRUCTURE analyses ΔK and mean LnK plots are presented in Online Resource 6. The ΔK of the STRUCTURE analysis of the entire dataset is three, yet with visual inspection on the mean LnK plot, K = 4 still increases the likelihood. In the species datasets the ΔK for DOM, HAM and PAL is two, confirmed by visual inspection of the mean LnK for DOM and PAL. For HAM, however, there is no large difference in mean likelihood for K = 1 (-5229.80) and K = 2 (-5132.83), thus not leading to a clear asymptotic shape in the mean LnK plot. Hence, we consider K = 1 to be the optimal number of genetic groups for HAM. Representative replicate barplots for the optimal K groups are in Fig. 5. In the full dataset (Fig. 5a) all individuals are assigned with great confidence to one of the four clusters, visible as each barplot having clearly more than 70% coloured in that of one genetic group, viz. M. domingensis (yellow), M. hamorii (pink) or one of the two populations of M. pallescens (green). For the analyses per species, M. domingensis was further subdivided in two populations (Fig. 5b), which roughly followed the two localities (i.e., BAR and ROD), yet 8 individuals had an unclear assignment or were assigned to the locality where they were not sampled (see Online Resource 7). The 8 trees that were assigned to the other locality in more than 70% of the runs have a DBH ranging from 4.7 to 19.9.
Figure 6 visualizes the DAPC analysis of the full dataset. The find.clusters function for this dataset showed that the lowest, significant drop in BIC score was at K = 4. Cross-validation for K = 4, resulted in 40 PCAs being retained for the DAPC analyses, in combination with DA = 3. The first axis shows a clear distinction between BAH and the three remaining populations, which are subsequently separated by the second axis. The results from the three other DAPC analyses can be found in Online Resource 7. Considering all four DAPC analyses, multiple, unambiguous genetic clusters were obtained for the full dataset and for PAL.
Pairwise FST, GST, G’ST and Jost’s D (DJ) values can be found in Table 3 and Online Resource 8. Values between populations range from 0.081 to 0.271 for FST, from 0.042 to 0.158 for GST, from 0.149 to 0.550 for G’ST and from 0.081 to 0.378 for DJ. Confidence intervals (CIs) of the pairwise indices can be found in Online Resource 9.
Diversity statistics for each population, locality and microsatellite marker are compiled in Online Resource 10. The mean values for the populations and localities can be found in Table 4. One population and two localities, namely EBV, ENT and MON, showed significant deviations from HWP for seven, four and one out of 16 markers, respectively, which resulted in an FIS that was significantly deviating from zero. Based on the three different types of analyses for genetic structure, five populations were defined: BAR, ROD, BAH, EBV and VAL.

STRUCTURE barplots for the three Magnolia species of the Dominican Republic. a: Dataset comprising all three species, K = 3. b: STRUCTURE runs per species, plotted against the dataset comprising all three species. Magnolia domingensis (BAR and ROD localities), K = 2. Magnolia hamorii (CAC, COR, LAG and TON localities), K = 2. Magnolia pallescens (CAS, SAL, ENT, MON, GUA, RAN localities), K = 2. Abbreviations represent sampling localities or populations as defined in Table 1

DAPC (Discriminant Analysis of Principal Components) plot for the Magnolias of the Dominican Republic. Names follow the species name (if K = 1) or population ID (if K > 1) as defined in Table 1. Four clusters are visible across three species: DOM (Magnolia domingensis), BAH (M. hamorii), EBV and VAL. The latter two belong to PAL (M. pallescens)
Discussion
Potential Magnolia distributions across Hispaniola
The ensemble SDM predicted the habitat of Magnolia across Hispaniola with great accuracy, as evidenced by the high model evaluation statistics obtained. Moreover, all 66 additional occurrences from the sampling effort in 2021 (which were not included in model fitting) fell within highly suitable predicted areas. Similarly, herbarium occurrence data that were deemed too unprecise for the SDM analyses, were often situated in highly suitable habitats as well (green to dark green areas in Fig. 3).
Regions of high habitat suitability for the Magnolias of Hispaniola co-occur with the numerous mountain ranges on the islands, which are annotated in Fig. 1. While we cannot rule out the possibility that our genus-level approach lacks species-specific nuances in Magnolia habitat requirements, we recommend future expeditions in search for undocumented Magnolia populations, to specifically focus on the areas of highest predicted habitat suitability for surveys, summarized in the following sentences. Mountain ranges with the most promising areas of detecting new Magnolia populations in the Dominican Republic are the Cordillera Central, the Sierra de Bahoruco and the Sierra de Neiba (Figs. 1 and 3). Possible remnant populations could be found in the Cordillera Septentrional and the Sierra Martín García, an isolated extension of the Sierra de Neiba. For Haiti, areas of interest reside in the mountain ranges Massif de la Hotte and Massif de la Selle, the Haitian counterpart of the Sierra de Bahoruco. However, previous expeditions to the Massif de la Selle did not find any hitherto unknown Magnolia species (pers. comm. Joel Timyan, Société Audubon Haiti). This can be the result of the species no longer being present in this area due to the severe deforestation in the mountain range itself, exacerbated by its relative proximity to the capital Port-au-Prince. To a lesser degree, possible areas of interest can be found in the Massif du Nord, Montagnes Noires and Chaîne des Matheux in central and northern Haiti (Figs. 1 and 3). Due to an extensive history of deforestation (Hedges et al. 2018) and the absence of dark green patches of suitable habitat in northern Haiti (Fig. 3) it was deemed unlikely that Magnolia populations still occurred here.
Knowledge about the potential habitat to guide survey designs for rare species is one of the most valuable uses of distribution modelling in ecology and conservation (McCune 2016). It also provides policy makers and on-the-ground conservationists a guideline for spatial prioritisation of conservation efforts (Villero et al. 2017). Concretely, it can serve as the basis for reinforcement efforts and the designation of habitat corridors; and allows to search for the true or full extent of populations more efficiently (Williams et al. 2009). Habitat corridors have successfully been applied by Baruah et al. (2019), Adhikari et al. (2012) and Liu et al. (2018).
Conservation Units and Priorities.
By integrating the results of the SDM (Fig. 3, Online Resource 5) and conservation genetic analyses (Figs. 5 and 6; Table 3, Online Resource 6–9), we propose to treat the five genetic Magnolia populations of the DR (i.e. BAR, ROD, BAH, EBV and VAL) as three conservation units (CU), following the morphospecies of Howard (1948).
On the one hand, because no unthreatened, related Magnolia species were included in this research, we can only make statements about the genetic diversity being healthy, low or high when comparing the localities and populations with each other (Spielman et al. 2004; Väli et al. 2008). On the other hand, we can also compare with diversity statistics generated in other studies, however, to be treated with caution. The genetic diversity of the five genetic populations appears ‘ample’ compared to other conservation genetic studies on (threatened) Magnolia species (Chávez-Cortázar et al. 2021; Aldaba Núñez et al. 2021) and only one out of five populations shows genetic signatures of inbreeding, with a low deviation from zero (Table 4). Assuming resources are limited, investments should most urgently go to the conservation of M. domingensis and M. ekmanii (M. ekmanii was examined in Veltjen et al. (2019)), followed by M. pallescens and lastly M. hamorii, based on the genetic (Table 4) and occurrence data (Fig. 3, Online Resource 5).
For M. domingensis the STRUCTURE (Fig. 5b) and DAPC (Online Resource 7) analyses on the species-specific dataset indicate two populations with a few migrant/relict trees in each of the localities, while these analyses on the full dataset (Figs. 5a and 6) and the SDM map (Fig. 3) only predict one. Additionally, the genetic differentiation statistics indicate intermediate values (Table 3) and there is a notable number of private alleles that sets these localities apart (Table 4).The genetic diversity of BAR is average across the Magnolias of the DR (Table 4). In contrast, ROD exhibits the worst genetic health among the five genetic populations (Table 4), with the lowest values for any A-statistic (i.e. A-, AR- and AP-values). However, both M. domingensis populations have low and non-significant FIS-values. As ROD exhibits low genetic diversity, genetic substructuring is not pronounced and the localities are linked by suitable habitat, we consider M. domingensis to be one CU, whereby we recommend reversing the ongoing genetic differentiation and low genetic diversity in ROD. This approach allows genetic rescue effects (i.e., a decrease in population extinction probability owing to gene flow; Bell et al. (2019)) between these populations to negate a further decline in genetic diversity (Ingvarsson 2001; Whiteley et al. 2014). Interestingly, due to an increment in sample size compared to Veltjen et al. (2019), we no longer see LD for population ROD. This was attributed to a recent bottleneck and a subsequent lack of random mating to restore linked loci (Slatkin 2008). Now, we can elucidate that this was a random sampling error, but see Waples (2015).
By integrating the genetic structuring results of M. ekmanii from Veltjen et al. (2019) with the SDM analyses (Fig. 3), we see a similar pattern as described for M. domingensis, namely, the species being split genetically in two populations. Based on the results of the SDM-analyses, this disjunction is unexpected considering that both populations are connected by a mountain range with highly suitable potential habitat. Further sampling efforts in the area could resolve this issue. Like the M. domingensis case described above, we treat M. ekmanii as a single CU, considering its genetic substructure and lower genetic health. Noteworthy, new occurrences of M. ekmanii were found relatively recently in the region of Bois Pangnol (easternmost point of the M. ekmanii occurences on Fig. 1; pers. comm. Joel Timyan, Société Audubon Haiti), an isolated location given the geographic distance between other Magnolias in Hispaniola. As these were not included in the genetic analysis, we cannot make inferences regarding their genetic structure or health. We recommend including this new locality in future conservation genetic studies.
According to the STRUCTURE and DAPC analyses (Figs. 5 and 6, Online Resource 7), M. pallescens is divided in two genetic groups: a northern population (EBV) in Ébano Verde Scientific Reserve and a southern one (VAL) in Valle Nuevo National Park (Fig. 3). Between EBV and VAL, we see an FST-value of 0.147 and a DJ of 0.154 (Table 3), which are considered moderate genetic fixation and allelic differentiation, respectively, following the quantification of Hartl and Clark (1997). This indicates that gene flow between the two populations has been low or absent for a significant amount of evolutionary time, allowing the populations to vary in allelic composition and frequency. When we compare infraspecific FST- and DJ-values with other studies on island endemic populations of trees, we find similar results. For example, FST = 0.000–0.229 for Pinus caribaea var. bahamensis, an endemic from the Bahaman archipelago (Sanchez et al. 2014). Recently, new individuals have been found in the suitable habitat patch between EBV and VAL, Reserva Cientifica Las Neblinas (henceforth RCLN), a previously unknown population of Magnolia pallescens (Figs. 1 and 3). Three historical herbarium vouchers (García 1002 [JBSD], García 1184 [JBSD] and Veloz 3237 [JBSD]) already implied a (historical) presence of M. pallescens in this patch, thus bridging the two populations geographically. These vouchers were excluded from the SDM analysis because of their imprecise occurrence data. In terms of genetic diversity, the population summary statistics for EBV and VAL indicate average genetic health when compared with other Magnolia populations from the DR (Table 4). Notably, the only three significant FIS-values are found across all populations and localities all belong to Magnolia pallescens: population EBV and two out of the four localities within population VAL (ENT and MON). These values are low compared to values reported for some other Magnolia species e.g. M. nuevoleonensis – population SPE and PE: FIS = 0.583* and 0.749* (Chávez-Cortázar et al. 2021); M. stellata – population Asahidani: FIS = 0.233* (Tamaki et al. 2016); M. tamaulipana – population LM: FIS = 0.47* (García-Montes et al. 2022). Interestingly, although the EBV population has a significant FIS-value, no significant FIS-values are found for its two localities. One potential explanation could be that a Wahlund effect was artificially created by merging the two localities (De Meeûs 2018). The Wahlund effect reduces heterozygosity in a population due to population substructuring (Wahlund 1928). The opposite situation from EBV is visible in VAL. Here, the VAL population does not have a significant FIS-value, yet they are significant for two out of the four localities of VAL. We attribute this to unrepresentative sampling across a small spatial scale. In terms of the other genetic diversity statistics, both M. pallescens populations, EBV and VAL, have average values. When comparing these results with Veltjen et al. (2019), no substantial differences are visible, aside from the significant FIS-value for EBV. Because of the recently discovered intermediate population and the significant FIS-value of population EBV and localities ENT and MON (population VAL), we propose to treat M. pallescens as one CU and hence actively aim to reverse ongoing genetic differentiation between the two populations. .
Magnolia hamorii has only one genetic population, BAH, situated in the Natural Protected Landscape Miguel Domingo Fuerte. The population, which represents the species, is most genetically differentiated from any other population as evidenced by the DAPC plot (Fig. 6), the DJ-values (Table 3) and the high number of private alleles (APP; Table 4). Geographically, the population is also isolated the most, both in distance as well as occurring in a distinct different mountain chain, as compared to M. pallescens and M. domingensis, that reside in different parts of the same mountain chain. Many of the genetic diversity indices (A, HE) of M. hamorii are the highest among the Magnolias of the DR (Table 4). Its FIS-value of 0.047 (Table 3) is the second highest among all populations, but non-significant and unproblematic as such. We consider M. hamorii as one CU since the sampled localities have one continuous modelled suitable habitat (Fig. 3) and there is no detectable genetic substructure (Table 3; Figs. 5a and 6).
The pattern of limited gene flow, leading to high genetic structuring, and little inbreeding is similar to other Magnolia studies such as those of Aldaba Núñez et al. (2021) and Veltjen et al. (2019). Inbreeding is expected to be minimal as the flowers of Magnolia are protogynous (Thien 1974) and promote outcrossing (Tamaki et al. 2009). However, geitonogamy has been reported in other Magnolia species (Bernhardt and Thien 1987; Hirayama et al. 2005; Tamaki et al. 2008) and regardless of selfing, inbreeding patterns can occur from mating with relatives e.g. in small and/or fragmented populations. The strong inbreeding depression in the early life stages of trees ensures that adult plants result solely from outcrossing (Sorensen 1999), increasing outcrossing rates and maintaining genetic diversity (Petit and Hampe 2006). High genetic diversity is desirable as their sessile lifestyle allows evolution of locally adapted ecotypes. Additionally, genetic diversity is “stored” in their great longevity, allowing genetic variants to circulate a long time within the populations (Alberto et al. 2013). This allows for potential reinforcement of genetically degraded populations (Aitken et al. 2008).
The results we report on the genetic diversity and habitat availability strongly nuance the Red List assessment categories of the studied species (Rivers et al. 2016) and confirm publications such as Vellend and Geber (2005) and Rivers et al. (2014). A first example: M. ekmanii has a relatively large potential habitat in Massif de la Hotte, exceeding that of M. domingensis (Fig. 3), which has comparable low values for its genetic diversity (Table 4 and Veltjen et al. 2019), while both are considered Critically Endangered (Rivers et al. 2016; Wheeler 2015). Hence with this information, M. domingensis can be considered in more dire need of urgent conservation action than M. ekmanii. A second example: M. pallescens and M. hamorii are both considered as Endangered, yet, when aligning the geographic results and genetic results we see an inverse relationship. Magnolia pallescens has the highest (potential) habitat, yet average genetic diversity with inbreeding, while M. hamorii has a smaller (potential) habitat, yet with higher genetic diversity and no inbreeding. This second example stresses that nor the current (estimated) geographic extent and knowledge on population trends are indicative of the true genetic diversity of the species, which relies on demographic history over a longer time.
Guidelines for conservation management.
To ensure a sustainable future for the Magnolias of Hispaniola, we propose to centralize conservation strategies around three main actions: exploration, protection, and reinforcement. Preferably for all the Magnolia species of Hispaniola, yet, as indicated in the previous paragraph, when resources are limited, following the defined priorities based on integrated genetic and SDM data (i.e. 1) M. domingensis & M. ekmanii, 2) M. pallescens and 3) M. hamorii).
Further exploration of the contemporary distribution of the Magnolias of Hispaniola should use Fig. 3 as a guideline. We propose the following strategies to search for new Magnolia individuals/populations, with decreasing priority. Initially, the highly suitable areas between the populations of M. domingensis and M. ekmanii should be explored, followed by nearby highly suitable, mountainous areas in the Massif de la Hotte, Sierra de Bahoruco and Cordillera Central. Subsequently, a search for remnant populations of M. emarginata and/or M. domingensis in Massif du Nord and Montagnes Noires. Finally, areas with no prior indication of Magnolia populations such as Sierra de Neiba, Sierra Martín García and Chaîne des Matheux should be further explored. Already, an exploration between the populations of M. domingensis has already been set up (Ramón Castillo Torres, pers. comm.).
Protection aims to conserve and expand the species’ habitat, TMCFs. Where possible, the Magnolias’ attractive properties can be used as an argument to expand or form new protected areas. In other words, use their potential as flagship species to draw attention to unprotected areas on the one hand and their potential as umbrella species to conserve not only the Magnolias, but TMCFs and their diversity on the other hand. Given that an adequate amount of genetic diversity is available within the conservation units, the focus should be on the expansion of protected areas. Ideally, these areas should strive to connect forest fragments in the landscape and the different populations as such. These habitat corridors (Christie and Knowles 2015) are especially important for the populations of M. ekmanii and M. domingensis, should no natural corridor exist. When successful, gene flow between populations should increase genetic diversity. In the case of M. hamorii, its population is under threat by the expansion of coffee plantations and tourism by the government. The latter has largely been negated by the Fundación Progressio (pers. comm. Ramón Elías Castillo Torres, Fundación Progressio). The major conservation action for M. hamorii therefore constitutes reducing future threats to the population.
Although protection is considered the most important goal in the long term, reinforcement is essential to cross this gap during which sufficient protection is established. Reinforcement aims to preserve and enhance genetic diversity and population sizes. Our interpretation of “reinforcement” follows Seddon et al. (2014): “The release of an organism into an existing population of conspecifics to enhance population viability.” Since gene flow between populations is limited, illustrated by the lower genetic health of ROD and a significant inbreeding value in EBV, we propose to execute reinforcement efforts between populations (Brichieri-Colombi and Moehrenschlager 2016). Here, populations BAR for Magnolia domingensis and VAL and RCLN for Magnolia pallescens will function as source populations of genetic diversity and ROD and EBV as their respective receptive sink populations. The benefits of minimizing loss of genetic diversity due to inbreeding and fragmentation outweighs the undoing of potential local adaptations. We encourage conservation practitioners to raise seedlings in nurseries and plant them close/within the sink population as randomly distributed as possible. Moreover, new populations could be established in suitable Magnolia habitat in the vicinity of known populations, preferably in an area secure from large scale threats, such as the area between ROD and BAR (M. domingensis). Nurseries should avoid hybridization and potential subsequent outbreeding depression (Keller et al. 2000; Tamaki et al. 2017) at any cost. Propagation efforts in nurseries are ongoing for the Magnolias of the DR, with mixed results (Castillo et al. 2018). New M. pallescens individuals are being planted in degraded areas near VAL; and also in nearby gardens, green areas of businesses etc. around EBV by the goodwill of the people. In the latter case, we advise to use individuals from VAL and RCLN as source material. For M. hamorii and M. domingensis, 1690 and 130 new individuals, respectively, have recently been planted with more on the way (Ramón Castillo Torres, pers. comm.). We emphasize the importance of collecting seeds from multiple localities and from as many different trees as possible across a large spatial scale. This ensures representative subsampling of the genetic diversity within the conservation unit (Oldfield and Newton 2012). To ensure and maximally extrapolate success, we propose to instate a monitoring programme to keep track of the survival rates of the seedlings and mature trees. This allows a quick and swift respond to new threats (Godefroid et al. 2011).
Conclusion
The SDM analyses suggest that the potential distribution is restricted to the various mountain ranges on Hispaniola. We indicate the mountains of the Montagnes Noires as a priority area to search for more remnant populations of the highly endangered M. emarginata and M. domingensis in Haiti. The Sierra de Neiba is the most promising area, without prior known Magnolia occurrences, to search for new populations in the Dominican Republic. Highly suitable habitat is present between known populations of M. domingensis, M. ekmanii and M. pallescens. These patches are highlighted for future reinforcement efforts should no natural habitat corridor exist. The Magnolias of the DR consist of five genetic populations, representing three species. We recommend each species to be treated as its own conservation unit (CU). The pattern of little inbreeding for Caribbean Magnolia species seems to persist in our expanded sampling. Population “Ébano Verde” and localities “Entrance” and “Montellano” exhibit minimal, but significant, inbreeding. Out of the three DR Magnolia species, M. domingensis is highlighted as it contained the lowest genetic diversity, the smallest number of (known) individuals, the lowest number of sampling localities and the lowest amount of potential habitat in its vicinity. For conservation management, we propose to focus on three actions: exploration, protection, and reinforcement. The main recommendation includes protection and instatement of habitat corridors between populations of M. domingensis and M. ekmanii, whereby the SDM results will serve as a guideline for spatial prioritization.
Data availability
The authors confirm that the supporting data of these findings are available within the article and its supplementary materials.
Code availability
Not applicable.
References
Adhikari D, Barik SK, Upadhaya K (2012) Habitat distribution modelling for reintroduction of Ilex khasiana Purk., a critically endangered tree species of northeastern India. Ecol Eng 40:37–43. https://doi.org/10.1016/j.ecoleng.2011.12.004
Aitken SN, Yeaman S, Holliday JA, Wang T, Curtis-McLane S (2008) Adaptation, migration or extirpation: climate change outcomes for tree populations. Evol Appl 1:95–111. https://doi.org/10.1111/j.1752-4571.2007.00013.x
Alberto FJ, Aitken SN, Alía R et al (2013) Potential for evolutionary responses to climate change - evidence from tree populations. Glob Chang Biol 19:1645–1661. https://doi.org/10.1111/gcb.12181
Aldaba Núñez FA, Veltjen E, Martínez Salas EM, Samain MS (2021) Disentangling species delineation and guiding conservation of endangered magnolias in Veracruz. Mexico Plants 10:673. https://doi.org/10.3390/plants10040673
Allouche O, Tsoar A, Kadmon R (2006) Assessing the accuracy of species distribution models: prevalence, kappa and the true skill statistic (TSS). J Appl Ecol 43:1223–1232. https://doi.org/10.1111/j.1365-2664.2006.01214.x
Barbet-Massin M, Jiguet F, Albert CH, Thuiller W (2012) Selecting pseudo-absences for species distribution models: how, where and how many? Methods Ecol Evol 3:327–338. https://doi.org/10.1111/j.2041-210X.2011.00172.x
Baruah PS, Deka K, Lahkar L, Sarma B, Borthakur SK, Tanti B (2019) Habitat distribution modelling and reinforcement of Elaeocarpus serratus L. - A threatened tree species of Assam, India for improvement of its conservation status. Acta Ecol Sin 39:42–49. https://doi.org/10.1016/j.chnaes.2018.06.002
Bell DA, Robinson ZL, Funk WC, Fitzpatrick SW, Allendorf FW, Tallmon DA, Whiteley AR (2019) The exciting potential and remaining uncertainties of genetic rescue. Trends Ecol Evol 34:1070–1079. https://doi.org/10.1016/j.tree.2019.06.006
Bernhardt P, Thien LB (1987) Self-isolation and insect pollination in the primitive angiosperms: new evaluations of older hypotheses. Plant Syst Evol 156:159–176. https://doi.org/10.1007/BF00936071
Brichieri-Colombi TA, Moehrenschlager A (2016) Alignment of threat, effort, and perceived success in north american conservation translocations. Conserv Biol 30:1159–1172. https://doi.org/10.1111/cobi.12743
Broennimann O, Fitzpatrick MC, Pearman PB et al (2012) Measuring ecological niche overlap from occurrence and spatial environmental data. Glob Ecol Biogeogr 21:481–497. https://doi.org/10.1111/J.1466-8238.2011.00698.X
Bubb P, May I, Miles L, Sayer J (2004) Cloud forest agenda. UNEP-WCMC, Cambridge, UK. http://www.unep-wcmc.org/resources/publications/UNEP_WCMC_bio_series/20.htm
Bystriakova N, Peregrym M, Erkens RHJ, Bezsmertna O, Schneider H (2012) Sampling bias in geographic and environmental space and its effect on the predictive power of species distribution models. Syst Biodivers 10:305–315. https://doi.org/10.1080/14772000.2012.705357
Cano-Ortiz A, Musarella CM, Piñar Fuentes JC, Pinto Gomes CJ, Cano E (2016) Distribution patterns of endemic flora to define hotspots on Hispaniola. Syst Biodivers 14:261–275. https://doi.org/10.1080/14772000.2015.1135195
Cano-Ortiz A, Musarella CM, Piñar Fuentes JC, Bartolomé Esteban C, Quinto-Canas R, Pinto Gomes C, Río SD, Cano E (2017) Advances in the knowledge of the vegetation of hispaniola (Caribbean Central America). In: Sebata A (Ed) Vegetation. IntechOpen. https://doi.org/10.5772/intechopen.72090
Castillo RE (2016) Magnolia Society – 3rd International Magnolia Symposium - Cuba. In: Magnolia Soc. https://www.magnoliasociety.org/page-1813213. Accessed 3 Mar 2020
Castillo RE, Encarnación Y, Peguero B, Clase T, Gratzfeld J (2018) Plan de acción de conservación integrada de las Magnolias (Magnoliaceae) amenazadas de República Dominicana – Magnolia domingensis, M. hamorii y M. pallescens. Fundación PROGRESSIO y Jardín Botánico Nacional Dr. Rafael M. Moscoso, República Dominicana
Chávez-Cortázar A, Oyama K, Ochoa-Zavala M, Mata-Rosas M, Veltjen E, Samain MS, Quesada M (2021) Conservation genetics of relict tropical species of Magnolia (section Macrophylla). Conserv Genet 22:259–273. https://doi.org/10.1007/s10592-021-01334-5
Christie MR, Knowles LL (2015) Habitat corridors facilitate genetic resilience irrespective of species dispersal abilities or population sizes. Evol Appl 8:454–463. https://doi.org/10.1111/eva.12255
De Meeûs T (2018) Revisiting FIS, FST, Wahlund effects, and null alleles. J Hered 109:446–456. https://doi.org/10.1093/jhered/esx106
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Elith J, Leathwick JR (2009) Species distribution models: ecological explanation and prediction across space and time. Annu Rev Ecol Evol Syst 40:677–697. https://doi.org/10.1146/annurev.ecolsys.110308.120159
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Figlar RB, Nooteboom HP (2004) Notes on Magnoliaceae IV. BLUMEA 49:87–100. https://doi.org/10.3767/000651904X486214
Frankham R, Ballou JD, Briscoe DA (2015) Introduction to conservation genetics, 2nd edn. Cambridge University Press, Cambridge, UK
García-Montes MA, Cibrián-Jaramillo A, Reyes-Zepeda F, Chacón-Hernández JC, Vanoye-Eligio V, Sánchez-González A, Octavio-Aguilar P (2022) Magnolia tamaulipana: genetic evaluation shows high vulnerability in a narrow distribution. Int. J. Plant Sci. 183(2). https://doi.org/10.1086/717817
Godefroid S, Piazza C, Rossi G, (2011) How successful are plant species reintroductions? Biol Conserv 144:672–682. https://doi.org/10.1016/j.biocon.2010.10.003
Goudet J (2000) FSTAT (Version 1.2): a computer program to calculate F-statistics. J Hered 91:509–511. https://doi.org/10.1093/jhered/91.6.509
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:372. https://doi.org/10.2307/2532296
Guillera-Arroita G, Lahoz-Monfort JJ, Elith J, Gordon A, Kujala H, Lentini PE, McCarthy MA, Tingley R, Wintle BA (2015) Is my species distribution model fit for purpose? Matching data and models to applications. Glob Ecol Biogeogr 24:276–292. https://doi.org/10.1111/geb.12268
Guisan A, Thuiller W (2005) Predicting species distribution: offering more than simple habitat models. Ecol Lett 8:993–1009. https://doi.org/10.1111/j.1461-0248.2005.00792.x
Hamilton LS, Juvik JO, Scatena FN (1995) The Puerto Rico tropical cloud forest symposium: introduction and workshop synthesis. In: Tropical montane cloud forests ecological studies, 1st edn. Springer, New York, NY, pp 1–23
Hao T, Elith J, Lahoz-Monfort JJ, Guillera-Arroita G (2020) Testing whether ensemble modelling is advantageous for maximising predictive performance of species distribution models. Ecography 43:549–558. https://doi.org/10.1111/ecog.04890
Hartl D, Clark A (1997) Principles of population genetics, 3rd edn. Sinauer, Sunderland, USA
Hedges SB, Cohen WB, Timyan J, Yang Z (2018) Haiti’s biodiversity threatened by nearly complete loss of primary forest. Proc Natl Acad Sci U S A 115:11850–11855. https://doi.org/10.1073/pnas.1809753115
Hedrick PW (2005) A standardized genetic differentiation measure. Evol (NY) 59:1633–1638. https://doi.org/10.1111/j.0014-3820.2005.tb01814.x
Hirayama K, Ishida K, Tomaru N (2005) Effects of pollen shortage and self-pollination on seed production of an endangered tree, Magnolia stellata. Ann Bot 95:1009–1015. https://doi.org/10.1093/aob/mci107
Holleley CE, Geerts PG (2009) Multiplex manager 1.0: a cross-platform computer program that plans and optimizes multiplex PCR. Biotechniques 46:511–517. https://doi.org/10.2144/000113156
Howard RA (1948) The morphology and systematics of the West Indian Magnoliaceae. Bull Torre Bot Club 75(4):335–357
Ingvarsson PK (2001) Restoration of genetic variation lost - the genetic rescue hypothesis. Trends Ecol Evol 16:62–63. https://doi.org/10.1016/s0169-5347(00)02065-6
IUCN - SSC Species Conservation Planning Sub-Committee (2017) Guidelines for species conservation planning, Version 1. IUCN, Gland, Switzerland. https://doi.org/10.2305/IUCN.CH.2017.18.en
Jombart T, Ahmed I (2011) Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071. https://doi.org/10.1093/bioinformatics/btr521
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026. https://doi.org/10.1111/j.1365-294X.2008.03887.x
Kalinowski ST, Taper ML (2006) Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv Genet 7:991–995. https://doi.org/10.1007/s10592-006-9134-9
Karger DN, Conrad O, Böhner J, Kawohl T, Kreft H, Soria-Auza RW, Zimmermann NE, Linder HP, Kessler M (2017) Climatologies at high resolution for the earth’s land surface areas. Sci Data 4:1–20. https://doi.org/10.1038/sdata.2017.122
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Keenan K, Mcginnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Keller M, Kollmann J, Edwards PJ (2000) Genetic introgression from distant provenances reduces fitness in local weed populations. J Appl Ecol 37:647–659. https://doi.org/10.1046/j.1365-2664.2000.00517.x
Kramer AT, Havens K (2009) Plant conservation genetics in a changing world. Trends Plant Sci 14:599–607. https://doi.org/10.1016/j.tplants.2009.08.005
Larridon I, Walter HE, Guerrero PC, Duarte M, Cisternas MA, Hernández CP, Bauters K, Asselman P, Goetghebeur P, Samain MS (2015) An integrative approach to understanding the evolution and diversity of Copiapoa (Cactaceae), a threatened endemic chilean genus from the Atacama desert. Am J Bot 102:1506–1520. https://doi.org/10.3732/ajb.1500168
Liu C, Newell G, White M, Bennett AF (2018) Identifying wildlife corridors for the restoration of regional habitat connectivity: a multispecies approach and comparison of resistance surfaces. PLoS ONE 13:e0206071. https://doi.org/10.1371/journal.pone.0206071
Liu C, Newell G, White M (2019) The effect of sample size on the accuracy of species distribution models: considering both presences and pseudo-absences or background sites. Ecography 42:535–548. https://doi.org/10.1111/ecog.03188
Lobo JM, Jiménez-Valverde A, Hortal J (2010) The uncertain nature of absences and their importance in species distribution modelling. Ecography 33:103–114. https://doi.org/10.1111/j.1600-0587.2009.06039.x
López-Barrera F, Bonilla-Moheno M, Toledo-Aceves T (2017) Cloud forest restoration with a landscape approach. Agro Prod 10:29–36. http://www.colpos.mx/wb_pdf/Agroproductividad/2017/AGROPRODUCTIVIDAD_10-1-2017.pdf
Louis EJ, Dempster ER (1987) An exact test for Hardy-Weinberg and multiple alleles. Biometrics 43:811. https://doi.org/10.2307/2531534
Malhi Y, Gardner TA, Goldsmith GR, Silman MR, Zelazowski P (2014) Tropical forests in the Anthropocene. Annu Rev Environ Resour 39:125–159. https://doi.org/10.1146/ANNUREV-ENVIRON-030713-155141
Maunder M, Leiva A, Santiago-Valentín E, Stevenson DW, Acevedo-Rodríguez P, Meerow AW, Mejía M, Clubbe C, Francisco-Ortega J (2008) Plant conservation in the caribbean island biodiversity hotspot. Bot Rev 74:197–207. https://doi.org/10.1007/s12229-008-9007-7
McCune JL (2016) Species distribution models predict rare species occurrences despite significant effects of landscape context. J Appl Ecol 53:1871–1879. https://doi.org/10.1111/1365-2664.12702
Medina RF, Barbosa P, Christman M, Battisti A (2006) Number of individuals and molecular markers to use in genetic differentiation studies. Mol Ecol Notes 6:1010–1013. https://doi.org/10.1111/j.1471-8286.2006.01476.x
Mod HK, Scherrer D, Luoto M, Guisan A (2016) What we use is not what we know: environmental predictors in plant distribution models. J Veg Sci 27:1308–1322. https://doi.org/10.1111/jvs.12444
Naimi B, Araújo MB (2016) Sdm: a reproducible and extensible R platform for species distribution modelling. Ecography 39:368–375. https://doi.org/10.1111/ecog.01881
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 70:3321–3323. https://doi.org/10.1073/pnas.70.12.3321
Nei M, Chesser RK (1983) Estimation of fixation indices and gene diversities. Ann Hum Genet 47:253–259. https://doi.org/10.1111/j.1469-1809.1983.tb00993.x
Nowshin S (2022) Magnolia species lost to science for 97 years rediscovered in Haiti. The Guardian, 3/8/2022. www.theguardian.com/environment/2022/aug/03/magnolia-species-lost-to-science-for-97-years-rediscovered-in-haiti
Oldfield S, Newton AC (2012) Integrated conservation of tree species by botanic gardens: a reference manual. Botanic Gardens Conservation International, Richmond, UK
Peakall R, Smouse PE (2006) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research – an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Petit RJ, Hampe A (2006) Some evolutionary consequences of being a tree. Annu Rev Ecol Evol Syst 37:187–214. https://doi.org/10.1146/annurev.ecolsys.37.091305.110215
Phillips SJ, Dudík M, Elith J, Graham CH, Lehmann A, Leathwick J, Ferrier S (2009) Sample selection bias and presence-only distribution models: implications for background and pseudo-absence data. Ecol Appl 19:181–197. https://doi.org/10.1890/07-2153.1
Phillips ND, Reid N, Thys T, Harrod C, Payne NL, Morgan CA, White HJ, Porter S, Houghton JDR (2017) Applying species distribution modelling to a data poor, pelagic fish complex: the ocean sunfishes. J Biogeogr 44:2176–2187. https://doi.org/10.1111/JBI.13033
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genet Soc Am 155:946–959. https://doi.org/10.1007/s10681-008-9788-0
R Core Team (2020) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/
Raymond M, Rousset F (1995) GENEPOP (Version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249. https://doi.org/10.1093/oxfordjournals.jhered.a111573
Rivers MC, Brummitt NA, Lughadha EN, Meagher TR (2014) Do species conservation assessments capture genetic diversity? Glob Ecol Conserv 2:81–87. https://doi.org/10.1016/j.gecco.2014.08.005
Rivers M, Beech E, Murphy L, Oldfield S (2016) The red list of Magnoliaceae - revised and extended. Botanic Gardens Conservation International, Surrey, UK
Roberge J, Angelstam P (2004) Usefulness of the umbrella species concept. Conserv Biol 18:76–85. https://doi.org/10.1111/j.1523-1739.2004.00450.x
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Sanchez M, Ingrouille MJ, Cowan RS, Hamilton MA, Fay MF (2014) Spatial structure and genetic diversity of natural populations of the Caribbean pine, Pinus caribaea var. bahamensis (Pinaceae), in the Bahaman archipelago. Bot J Linn Soc 174:359–383. https://doi.org/10.1111/boj.12146
Sangermano F, Bol L, Galvis P, Gullison RE, Hardner J, Ross GS (2015a) Habitat suitability and protection status of four species of amphibians in the Dominican Republic. Appl Geogr 63:55–65. https://doi.org/10.1016/j.apgeog.2015.06.002
Sangermano F, Bol L, Galvis P, Gullison RE, Hardner J, Ross GS (2015b) Forest baseline and deforestation map of the Dominican Republic through the analysis of time series of MODIS data. Data Br 4:363–367. https://doi.org/10.1016/j.dib.2015.06.006
Seddon PJ, Griffiths CJ, Soorae PS, Armstrong DP (2014) Reversing defaunation: restoring species in a changing world. Science 345:406–412. https://doi.org/10.1126/science.1251818
Slatkin M (2008) Linkage disequilibrium - understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9:477–485. https://doi.org/10.1038/nrg2361
Sorensen FC (1999) Relationship between self-fertility, allocation of growth, and inbreeding depression in three coniferous species. Evol (NY) 53:417–425. https://doi.org/10.1111/j.1558-5646.1999.tb03777.x
Spielman D, Brook BW, Frankham R (2004) Most species are not driven to extinction before genetic factors impact them. Proc Natl Acad Sci U S A 101:15261–15264. https://doi.org/10.1073/pnas.0403809101
Stas M, Aerts R, Hendrickx M et al (2020) An evaluation of species distribution models to estimate tree diversity at genus level in a heterogeneous urban-rural landscape. Landsc Urban Plan 198. https://doi.org/10.1016/J.LANDURBPLAN.2020.103770
Tamaki I, Setsuko S, Tomaru N (2008) Genetic variation and differentiation in populations of a threatened tree, Magnolia stellata: factors influencing the level of within-population genetic variation. Heredity 100:415–423. https://doi.org/10.1038/sj.hdy.6801097
Tamaki I, Setsuko S, Tomaru N (2009) Estimation of outcrossing rates at hierarchical levels of fruits, individuals, populations and species in Magnolia stellata. Heredity 102:381–388. https://doi.org/10.1038/hdy.2008.128
Tamaki I, Setsuko S, Tomaru N (2016) Genetic diversity and structure of remnant Magnolia stellata populations affected by anthropogenic pressures and a conservation strategy for maintaining their current genetic diversity. Conserv Genet 17:715–725. https://doi.org/10.1007/s10592-016-0817-6
Tamaki I, Tani S, Setsuko S, Ueno S, Wadasaki N, Tomaru N (2017) Reduced incompatibility in the production of second generation hybrids between two Magnolia species revealed by bayesian gene dispersal modeling. Am J Bot 104:1546–1555. https://doi.org/10.3732/ajb.1700138
Thien LB (1974) Floral biology of Magnolia. Am J Bot 61:1037–1045. https://doi.org/10.2307/2441921
Thiers B (2022) Index Herbariorum: a global directory of public herbaria and associated staff. New York Botanical Garden’s Virtual Herbarium. http://sweetgum.nybg.org/science/ih/
Title PO, Bemmels JB (2018) ENVIREM: an expanded set of bioclimatic and topographic variables increases flexibility and improves performance of ecological niche modeling. Ecography 41:291–307. https://doi.org/10.1111/ecog.02880
Toledo-Aceves T, Trujillo-Miranda AL, López-Barrera F (2021) Tree regeneration in active and passive cloud forest restoration: functional groups and timber species. For Ecol Manage 489:119050. https://doi.org/10.1016/j.foreco.2021.119050
Väli Ü, Einarsson A, Waits L, Ellegren H (2008) To what extent do microsatellite markers reflect genome-wide genetic diversity in natural populations? Mol Ecol 17:3808–3817. https://doi.org/10.1111/j.1365-294X.2008.03876.x
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
VanDerWal J, Shoo LP, Graham C, Williams SE (2009) Selecting pseudo-absence data for presence-only distribution modeling: how far should you stray from what you know? Ecol Modell 220:589–594. https://doi.org/10.1016/j.ecolmodel.2008.11.010
Vartia S, Collins PC, Cross TF, Fitzgerald RD, Gauthier DT, McGinnity P, Mirimin L, Carlsson J (2014) Multiplexing with three-primer PCR for rapid and economical microsatellite validation. Hereditas 151:43–54. https://doi.org/10.1111/hrd2.00044
Vellend M, Geber MA (2005) Connections between species diversity and genetic diversity. Ecol Lett 8:767–781. https://doi.org/10.1111/j.1461-0248.2005.00775.x
Veltjen E, Asselman P, Hernández Rodríguez M, Palmarola Bejerano A, Testé Lozano E, González Torres LR, Goetghebeur P, Larridon I, Samain MS (2019) Genetic patterns in neotropical magnolias (Magnoliaceae) using de novo developed microsatellite markers. Heredity 122:485–500. https://doi.org/10.1038/s41437-018-0151-5
Veltjen E (2020) The Caribbean Magnolia species (Magnoliaceae): assessment of the genetic diversity and the underlying evolutionary history. PhD Dissertation, Ghent University, Belgium
Veltjen E, Testé E, Palmarola Bejerano AP, Asselman P, Hernández Rodríguez M, González Torres LR, Chatrou LW, Goetghebeur P, Larridon I, Samain MS (2022) The evolutionary history of the caribbean magnolias (Magnoliaceae): testing species delimitations and biogeographical hypotheses using molecular data. Mol Phylogenet Evol 167:107359. https://doi.org/10.1016/J.YMPEV.2021.107359
Venter O, Sanderson EW, Magrach A, Allan JR, Beher J, Jones KR, Possingham HP, Laurance WF, Wood P, Fekete BM, Levy MA, Watson JE (2016) Global terrestrial human footprint maps for 1993 and 2009. Sci Data 3:1–10. https://doi.org/10.1038/sdata.2016.67
Venter O, Sanderson EW, Magrach A, Allan JR, Beher J, Jones KR, Possingham HP, Laurance WF, Wood P, Fekete BM, Levy MA, Watson JE (2018) Last of the wild project, version 3 (LWP-3): 2009 human footprint, 2018 release. NASA socioeconomic Data and, Palisades, New York. https://doi.org/10.7927/H46T0JQ4. Applications Center (SEDAC)
Villero D, Pla M, Camps D, Ruiz-Olmo J, Brotons L (2017) Integrating species distribution modelling into decision-making to inform conservation actions. Biodivers Conserv 26:251–271. https://doi.org/10.1007/s10531-016-1243-2
Wahlund S (1928) Zusammensetzung von Populationen und Korrelationerscheinungen vom Standpunkt der Vererbungslehre aus betrachtet. Hereditas 11:65–106. https://doi.org/10.1111/j.1601-5223.1928.tb02483.x
Wang H, Yang B, Wang H, Xiao H (2021) Impact of different numbers of microsatellite markers on population genetic results using SLAF-seq data for Rhododendron species. Sci Rep 11:1–9. https://doi.org/10.1038/s41598-021-87945-x
Waples RS (2015) Testing for Hardy–Weinberg proportions: have we lost the plot? J Hered 106:1–19. https://doi.org/10.1093/jhered/esu062
Ward SM, Jasieniuk M (2009) Review: sampling weedy and invasive plant populations for genetic diversity analysis. Weed Sci 57:593–602. https://doi.org/10.1614/ws-09-082.1
Warren DL, Glor RE, Turelli M (2010) ENMTools: a toolbox for comparative studies of environmental niche models. Ecography 33:607–611. https://doi.org/10.1111/j.1600-0587.2009.06142.x
Weckworth BV, Hebblewhite M, Mariani S, Musiani M (2018) Lines on a map: conservation units, meta-population dynamics, and recovery of woodland caribou in Canada. Ecosphere 9(7):e02323. https://doi.org/10.1002/ecs2.2323
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370. https://doi.org/10.2307/2408641
Wheeler L (2015) Magnolia domingensis. In: IUCN Red List Threat Species 2015 eT193937A2291464. https://doi.org/10.2305/IUCN.UK.2015-2.RLTS.T193937A2291464.en. Accessed 20 May 2021
Whiteley AR, Fitzpatrick SW, Funk WC, Tallmon DA (2014) Genetic rescue to the rescue. Trends Ecol Evol 30:42–49. https://doi.org/10.1016/j.tree.2014.10.009
Williams JN, Seo C, Thorne J, Nelson JK, Erwin S, O’Brien JM, Schwartz MW (2009) Using species distribution models to predict new occurrences for rare plants. Divers Distrib 15:565–576. https://doi.org/10.1111/j.1472-4642.2009.00567.x
Wilson SJ, Rhemtulla JM (2018) Small montane cloud forest fragments are important for conserving tree diversity in the ecuadorian Andes. Biotropica 50:586–597. https://doi.org/10.1111/BTP.12542
Zurell D (2020) Introduction to species distribution modelling (SDM) in R. https://damariszurell.github.io/SDM-Intro/. Accessed 24 Mar 2020
Acknowledgements
We are grateful to the authorities of the Dominican Republic and Haiti for issuing the permits DQCSPAP/SIC/365 and VAPB-01488, respectively. We thank Andy Vierstraete, Pieter Asselman and Ana María Rueda Urrego for help in the lab. Also, we thank Rosa Rodríguez Peña, Majela Hernández Rodríguez, Víctor Martín Vélez, Jean-Francois Orilién Beauduy, William Cinéa, Joel Timyan and Roland Trézil for their help with the fieldwork. Finally, we thank Thibeaut Formesyn for his help with the QGIS map production.
Funding
We thank the Fondation Franklinia (2019-03), Fundación PROGRESSIO, Ghent University (BOF.DOC.2014.0062.01) and the Research Foundation Flanders (V4.453.15 N) for their financial support.
Author information
Authors and Affiliations
Contributions
Conceptualization: Emily Veltjen, Marie-Stéphanie Samain; Methodology: Diederik Strubbe, Emily Veltjen; Formal analysis and investigation: Tim Claerhout, Emily Veltjen; Writing – original draft preparation: Tim Claerhout; Writing – review and editing: Tim Claerhout, Diederik Strubbe, Ramón Elías Castillo Torres, Marie-Stéphanie Samain, Lars Chatrou, Emily Veltjen; Funding acquisition: Emily Veltjen, Marie-Stéphanie Samain; Resources: Emily Veltjen, Ramón Elías Castillo Torres; Supervision: Diederik Strubbe, Emily Veltjen, Lars Chatrou.
Corresponding author
Ethics declarations
Conflict of interest/competing interests
The authors hereby declare no conflict of interests.
Ethics approval
Not applicable.
Consent to participate
All authors hereby consent to participate.
Consent for publication
All authors hereby consent for publication.
Additional information
Communicated by Daniel Sanchez Mata.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Claerhout, T., Strubbe, D., Castillo Torres, R. et al. An integrative study of species distribution modelling and conservation genetics: Magnolia in Hispaniola. Biodivers Conserv 32, 1205–1231 (2023). https://doi.org/10.1007/s10531-023-02546-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-023-02546-6