
Abstract
Early detection and identification of plant pathogens is one of the most important strategies for sustainable plant disease management. Fast, sensitive, and accurate methods that are cost-effective are crucial for plant disease control decision-making processes. Coffee leaf rust (CLR) caused by Hemileia vastatrix is a devastating worldwide fungal disease which causes serious yield losses of coffee, especially relevant for Coffea arabica. A rapid PCR assay for detecting and characterizing H. vastatrix with high specificity, high sensitivity and simple operation has been developed based on specific amplification of the Internal Transcribed Spacer (ITS) region of ribosomal genes. The specificity of the primers was determined using isolates DNA of H. vastatrix, Coleosporium plumeriae, and other fungal species that infect coffee plants and are common in coffee leaves, such as Lecanicillium sp (the H. vastatrix hyperparasite fungi), Cercospora coffeicola, Colletotrichum gloeosporioides, amongst others. Results showed specific amplification of a 396-bp band from H. vastatrix DNA with a detection limit of 10 pg/μl of pure genomic DNA of the pathogen. The PCR assay described in the current chapter allows to detect H. vastatrix rapidly and reliably in naturally infected coffee tissues, vital for the early detection and diagnostics of H. vastatrix and CLR epidemiology.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Coffee
- Hemileia vastatrix
- Early molecular detection
- Polymerase chain reaction
- Internal transcribed spacer
- Specificity and sensitivity
1 Introduction
Accurate identification and diagnosis of plant diseases are vital for prevention of the spread of invasive pathogens (Balodi et al. 2017). So far, advances in the development of molecular methods have provided diagnostic laboratories with powerful tools for the detection and identification of phytopathogens, among which polymerase chain reaction (PCR) and other DNA-based techniques proved to be rapid and highly suitable approaches to improve the accuracy and efficiency of plant pathogen detection and characterization (Lévesque et al. 1998; Haudenshield et al. 2017). Detection protocols used for the diagnosis or quarantine measures should be reproducible and cost effective, time saving and simple in procedure (Elnifro et al. 2000; Hayden et al. 2008; Tomkowiak et al. 2019). In addition, sensitivity to pathogen concentration, and specificity to genetic variability within a target pathogen population are also high priorities for molecular detection (Balodi et al. 2017).
The Internal Transcribed Spacer (ITS) of the ribosomal DNA show high inter-species variability and intra-species stability and conservation, and hence is considered a reliable DNA marker to identify and classify the pathogenic fungi (Glynn et al. 2010). PCR assays based on the ITS region have been widely used for the detection of fungal pathogens in different crops such as sunflower, tobacco, soybean, cedar trees, miscanthus and others (Guglielmo et al. 2007; Chen et al. 2008; Torres-Calzada et al. 2011; Capote et al. 2012), relating to the pathogens of Phytophthora (Grünwald et al. 2012; Patel et al. 2016), Puccinia (Guo et al. 2016), Verticillium spp. (Nazar et al. 1991), Pleurotus spp. (Ma and Luo 2002), Pyricularia and anthracnose (Sugawara et al. 2009), Saccharomyces saccharum (Anggraini et al. 2019), Podosphaera xanthii (Tsay et al. 2011) and Golovinomyces cichoracearum (Troisi et al. 2010). This technique was applied to differentiate two pathotypes of Verticillium alboatrum infecting hop, to distinguish 11 taxons of wood decay fungi infecting hardwood trees, and to differentiate multiple Phytophthora species from plant material and environmental samples (Shamim et al. 2017; Belete and Boyraz 2019).
Coffee leaf rust (CLR), a major disease of Arabica coffee (Coffea arabica L.), is caused by the obligate biotrophic fungus Hemileia vastatrix Berkeley and Broome (Talhinhas et al. 2017). The infection of coffee leaves by H. vastatrix starts with urediniospore germination, appressorium formation over stomata, penetration, and inter- and intracellular colonization without any visible symptoms in the early stages of the infection in the field conditions < 10 days (Talhinhas et al. 2017; Silva et al. 2018). In field conditions, the visible rust spores can be observed about 20 days after the first infection of H. vastatrix (Schieber 1972). So far, the traditional method for detecting and characterizing CLR was time-consuming and laborious, and relied on conventional morphological examination requiring professional taxonomic knowledge and extensive experience (McCartney et al. 2003; Silva et al. 2012). Hence, rapid and high-throughput identification and detection methods for H. vastatrix are required to recognize the infection as early as possible before the appearance and spread of CLR spores in the leaf surface. Early detection methods can facilitate implementing proper management approaches to prevent the development and spread of the coffee leaf rust pathogen (Sankaran et al. 2010).
The present study was undertaken with the objective of early detection of H. vastatrix based on the PCR amplification of a specific ITS region in the rDNA of H. vastatrix. A simple, accurate and rapid PCR-based assay for CLR is presented as a reliable technique to monitor H. vastatrix in the early stages of the infection, as well as to provide scientific basis for the prevention and control of CLR.
2 Materials
-
1.
ddH2O.
-
2.
1 X TE buffer (pH 8.0).
-
3.
CTAB.
-
4.
KAc.
-
5.
Chloroform.
-
6.
Isoamyl alcohol.
-
7.
Isopropanol.
-
8.
75% ethanol.
-
9.
Anhydrous ethanol.
-
10.
Phenol.
-
11.
Na2Ac.
-
12.
rTaq (Dalian TaKaRa Co., Ltd., 5 U/µl).
-
13.
10X PCR Buffer (Mg2+ plus).
-
14.
dNTPs (2.5 mM).
-
15.
Biowest regular agarose G-10 (CB005-100G).
-
16.
Tris/borate electrophoresis buffer.
-
17.
Microwave.
-
18.
GoldView II Nuclear Staining Dyes (5,000×) (Solarbio® LIFE SCIENCES).
-
19.
Electrophoresis tank.
-
20.
DL 2000 Marker (Dalian TaKaRa Co., Ltd.).
-
21.
RNAse A solution (Solarbio® LIFE SCIENCES, 10 mg/ml).
-
22.
Water bath.
-
23.
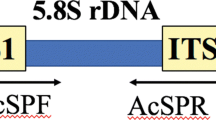
Specific primers (see Fig. 1).
Fig. 1 
Primers Hv-ITS-F/R designed for H. vastatrix PCR assay based on rDNA-ITS sequences
-
24.
Genomic DNA of the pathogen (see Fig. 2).
Fig. 2 
Example of specificity test of Hv-ITS-F/R primer sets. The DNA of 4 strains of H. vastatrix (lanes 2–5), 8 other fungi (lanes 6–13) (see Note 5) and sterilized ddH2O as the negative control (lane 1) were amplified by PCR using Hv-ITS-F/R primers. Primers for Hv-ITS-F/R amplify a 396-bp specific band from the DNA of H. vastatrix, while no bands were observed from the DNA of other fungi. M: DL 2000 DNA marker; 1: ddH2O control; 2–5: H. vastatrix; 6: Colletotrichum gloeosporioides; 7: Lecanicillium sp.; 8: Cercospora coffeicola; 9: Coleosporium plumeriae; 10: Colletotrichum falcatum; 11: Ustilago scitaminea; 12: Leptosphaeria sacchari; 13: Aspergillus niger
-
25.
Ice.
-
26.
Ice machine.
-
27.
Autoclave.
-
28.
Mortar.
-
29.
Measuring cylinder (100 ml).
-
30.
Scissors.
-
31.
Liquid nitrogen.
-
32.
Micropipette (1,000, 200, 10, 2.5 μl).
-
33.
Centrifuge tube (1.5, 2 ml).
-
34.
NanoDrop 2000c Spectrophotometer (Thermo Scientific, USA).
-
35.
PTC-100™ Programmable Thermal Controller (MJ Research Inc, USA).
-
36.
BIO-RAD GelDoc 2000 GelDoc 2000™.
-
37.
Power/PAC300.
-
38.
PCR tubes (0.2 ml).
-
39.
Tips (1,000, 200, 10 µl).
-
40.
Absolute alcohol.
-
41.
Refrigerated Centrifuge Sigma 3k15.
-
42.
SCILOGEX_D2012_Centrifuge.
-
43.
Refrigerator.


3 Methods
3.1 Designing the Specific Primers for Hemileia vastatrix
-
1.
The primers Hv-ITS-F/R were designed to specifically amplify the ITS2 region of H. vastatrix. The sequence of the forward primer Hv-ITS-F is 5’-GGTACACCTGTTTGAGAGTATG-3’, and the sequence of the reverse primer is Hv-ITS-R is 5’-CAAAATATGTCATACCTCTCATTCT-3 (see Fig. 1).
-
2.
Primer sequences of Hv-ITS-F and Hv-ITS-R were used as inputs for a BLAST search against the NCBI database to confirm the specificity. The primers were synthesized by Invitrogen Biotechnology (Shanghai) Co., Ltd.
-
3.
Upon delivery, dilute lyophilized primers to the concentration of 10 µM by adding 0.1 X TE buffer. Store at − 20 °C for later use.
3.2 Total DNA Extraction from Suspected Diseased Leaves or Typical Diseased Samples
The CTAB method (Siegel et al. 2017) was used to extract DNA from diseased leaves.
-
1.
Preheat the CTAB extraction buffer to 65 °C in a water bath.
-
2.
Grind approximately 1 g of diseased leaf tissue into a fine powder in a mortar using liquid nitrogen (see Note 1).
-
3.
Add 15 ml of pre-heated CTAB buffer into each tube. Mix well and incubate at 65 °C for 30 min. Turn the tubes upside down every 10 min to resuspend the samples in the buffer (see Note 2).
-
4.
Add 3 ml of 5 M KAc to the tube containing the lysate and let it stand on ice for 20 min.
-
5.
Add the same volume of a chloroform:iso-amyl alcohol (24:1) mixture to the tube, mix well and centrifuge at 12,000 rpm at 4 °C for 15 min.
-
6.
Repeat step 4.
-
7.
After centrifugation, transfer supernatant into a new tube.
-
8.
Add 12 ml of a pre-cooled isopropanol, mix by inverting and put at −20 °C to fully precipitate the DNA.
-
9.
Centrifuge the tube at 10,000 rpm for 15 min to pellet the DNA.
-
10.
Rinse the pellet twice with 75% ethanol, and once with anhydrous ethanol. Air-dry the DNA pellet and dissolve in 10 ml TE buffer.
-
11.
Treat the DNA samples with 1 μl RNase (10 mg/ml) at room temperature for 1–2 h.
-
12.
Add the same volume of phenol: chloroform: isoamyl alcohol (25:24:1), mix well and then centrifuge at 12,000 rpm at 4 °C for 15 min.
-
13.
Transfer the supernatant to a new tube, mix with 1 ml ice-cold 3 M Na2Ac, and 20 ml of anhydrous ethanol, and place at − 20 °C overnight.
-
14.
Centrifuge at 12,000 rpm for 30 min at 4 °C.
-
15.
Discard the supernatant, rinse the DNA pellet with 75% ethanol and dissolve in 1 ml TE after drying.
-
16.
Determine the DNA concentration by e.g., a NanoDrop 2000c.
-
17.
Store the DNA at − 20 °C until further use (see Note 3).
3.3 Preparation of the PCR Reaction Mixture and PCR Amplification
-
1.
Prepare a 20 µl PCR reaction mix as follows (see Note 4):
10X PCR Buffer | 2 µl |
dNTPs (2.5 mM) | 1.6 µl |
Forward Primer (10 µM) | 1 µl |
Reverse Primer (10 µM) | 1 µl |
rTaq (5 U/µl) | 0.1 µl |
DNA template | 1 µl |
ddH2O | 14.2 µl |
-
2.
Mix all components, spin briefly and immediately place in a thermocycler (here a gradient Mastercycler was used).
-
3.
Set the thermocycler conditions as following: initial denaturation at 94 °C for 3 min, denaturation at 94 °C for 30 s, annealing at 62 °C for 30 s, extension at 72 °C for 1 min, 35 cycles; final extension time at 72 °C for 5 min.
-
4.
Upon termination store samples at 15 °C.
3.4 Gel Electrophoresis
-
1.
Prepare a 1% agarose gel by mixing 1 g of agarose and 100 ml of TBE buffer (pH 8.0).
-
2.
Melt thoroughly in a microwave.
-
3.
Allow the mixture to cool down to 40 °C, add 1 µl GoldView DNA dye solution (1 µl/100 ml gel) and mix. Pour the gel and allow to solidify.
-
4.
Load 10 μl of PCR products and run at 120 V for 20 min.
-
5.
View the gel under the UV light. The H. vastatrix positive samples are defined as the ones that show a specific single band of 396-bp (see Figs. 2 and 3).
Fig. 3 
Example sensitivity test of primer sets Hv-ITS-F/R. Prepare a series of DNA concentrations to determine the sensitivity of the detection system. The initial genomic DNA concentration of H. vastatrix was adjusted to 10 ng/μL, with serial tenfold dilutions to reach 10−5 ng/μl. The results showed that samples with DNA concentration of 10 pg/μL or higher yielded a clearly visible 396-bp band while samples with a lower concentration were negative. M: DL 2 000 DNA marker; 1: ddH2O control; 2: 10 ng/μl; 3: 1 ng/μl; 4: 10−1 ng/μl; 5: 10−2 ng/μl; 6: 10−3 ng/μl; 7: 10−4 ng/μl; 8: 10−5 ng/μl

4 Notes
-
1.
The leaf samples should be fully ground into a fine powder. To prevent sample cross-contamination, change gloves after finishing each sample.
-
2.
All the tubes, tips and utensils should be sterilized prior to use.
-
3.
To prevent cross contamination, the pipette tips must be used once after contact with samples.
-
4.
The PCR reaction mix is prepared on ice in a clean environment.
-
5.
The urediniospores of Coleosporium plumeriae and the other fungal isolates were extracted using a Fungal DNA kit (E.Z.N.A.TM Fungal DNA Kit, Omega, Bio-tek, USA) according to the manufacturer’s protocol.
References
Anggraini I, Ferniah RS, Kusdiyantini DE (2019) Isolasi khamir fermentatif dari batang tanaman tebu (Saccharum officinarum. L) dan hasil identifikasinya berdasarkan sekuens internal transcribed spacer. Berkala bioteknologi 2(2):12–22
Balodi R, Bisht S, Ghatak A, Rao KH (2017) Plant disease diagnosis: technological advancements and challenges. Indian Phytopathol 70(3):275–281
Belete T, Boyraz N (2019) Biotechnological tools for detection, identification and management of plant diseases. Afr J Biotechnol 18(29):797–807
Capote N, Pastrana AM, Aguado A, Sánchez-Torres P (2012) Molecular tools for detection of plant pathogenic fungi and fungicide resistance. Plant Pathol 151–202
Chen RS, Huang CC, Li JC, Tsay JG (2008) First report of Simplicillium lanosoniveum causing brown spot on Salvinia auriculata and S. molesta in Taiwan. Plant Dis 92(11):1589–1589
Elnifro EM, Ashshi AM, Cooper RJ, Klapper PE (2000) Multiplex PCR: optimization and application in diagnostic Virology. Clin Microbiol Rev 13(4):559–570
Glynn NC, Dixon LJ, Castlebury LA, Szaboc LJ, Comstock JC (2010) PCR assays for the sugarcane rust pathogens Puccinia kuehnii and P. Melanocephala and detection of a SNP associated with geographical distribution in P. kuehnii. Plant Pathol 59(4):703–711
Grünwald NJ, Werres S, Goss EM, Taylor CR, Fieland VJ (2012) Phytophthora obscura sp. nov., a new species of the novel Phytophthora subclade 8d. Plant Pathol 61:610–622
Guglielmo F, Bergemann SE, Gonthier P, Nicolotti G (2007) A multiplex PCR-based method for the detection and early identification of wood rotting fungi in standing trees. J Appl Microbiol 103(5):1490–1507
Guo DD, Zhao SH, Zhang HZ, Zhao J, Jing L (2016) Fungicides screening for management of sunflower rust caused by Puccinia henlianthi. Chin J Oil Crop Sci 38(3):388–394
Haudenshield JS, Song JY, Hartman GL (2017) A novel, multiplexed, probe-based quantitative PCR assay for the soybean root- and stem-rot pathogen, Phytophthora sojae, utilizes its transposable element. PLoS ONE 12(4):e0176567
Hayden MJ, Nguyen T, Waterman A, Chalmers KJ (2008) Multiplex-ready PCR: a new method for multiplexed SSR and SNP genotyping. BMC Genomics 9(1):80–80
Lévesque CA, Harlton CE, de Cock AWAM (1998) Identification of some oomycetes by reverse dot blot hybridization. Phytopathology 88:213–222
Ma FY, Luo XC (2002) Phylogeny of Pleurotus inferred from PCR-RFLP of 28S ribosomal DNA. J Huazhong (Cent China) Agric Univ 21(3):201–205
McCartney HA, Foster SJ, Fraaije BA, Ward E (2003) Molecular diagnostics for fungal plant pathogens. Pest Manag Sci 59(2):129–142
Nazar RN, Hu X, Schmidt J, Culham D (1991) Potential use of PCR-amplified ribosomal intergenic sequences in the detection and differentiation of verticillium wilt pathogens. Physiol Mol Plant Pathol 39(1):1–11
Patel JS, Vitoreli A, Palmateer AJ, El-Sayed A, Noman DJ, Goss EM, Brennan MS, Ali GS (2016) Characterization of Phytophthora spp. Isolated from ornamental plants in Florida. Plant Dis 100:500–509
Sankaran S, Mishra A, Ehsani R, Davis C (2010) A review of advanced techniques for detecting plant diseases. Comput Electron Agric 72(1):1–13
Schieber E (1972) Leaf blight incited by Ascochyta coffeae on coffee in Guatemala. Plant Dis Reporter 56(9):753–754
Shamim M, Kumar P, Kumar RR, Kumar M, Kumar RR, Singh KN (2017) Assessing fungal biodiversity using molecular markers. Mol Markers Mycol 15:305–333. https://doi.org/10.1007/978-3-319-34106-4
Siegel CS, Stevenson FO, Zimmer EA (2017) Evaluation and comparison of FTA card and CTAB DNA extraction methods for non-agricultural taxa. Appl Plant Sci 5(2):1600109
Silva AVC, Santos ARF, Lédo AS, Feitosa RB, Almeida CS, Silva GM, Rangel MSA (2012) Moringa genetic diversity from germplasm bank using RAPD Markers. Trop Subtrop Agroecosyst 15:31–39
Silva DN, Varzea V, Paulo OS, Batista D (2018) Population genomic footprints of host adaptation, introgression and recombination in coffee leaf rust. Mol Plant Pathol 19(7):1742–1753
Sugawara K, Matsudate A, Ito Y, Nanai T (2009) Anthracnose of Christmas rose caused by Colletotrichum sp. J Gen Plant Pathol 75(2):163–166
Talhinhas P, Batieta D, Diniz I, Vieira A, Sliva DN, Loureiro A, Tavares S, Pereira AP, Azinheira HG, Guerra-Guimarães L, Várzea V, Silva MDC (2017) The coffee leaf rust pathogen Hemileia vastatrix: one and a half centuries around tropics. Mol Plant Pathol 18(8):1039–1051
Tomkowiak A, Bocianowski J, Radzikowska D, Kowalczewski PL (2019) Selection of parental material to maximize heterosis using SNP and SilicoDarT markers in maize. Plants 8:349
Torres-Calzada C, Tapia-Tussell R, Quijano-Ramayo A, Martin-Mex R, Rojas-Herrera R, Higuera-Ciapara I, Perez-Brito D (2011) A Species-specific polymerase chain reaction assay for rapid and sensitive detection of Colletotrichum capsici. Mol Biotechnol 49(1):48–55
Troisi M, Bertetti D, Garibaldi A, Gullino ML (2010) First report of powdery mildew caused by Golovinomyces cichoracearum on Gerbera (Gerbera jamesonii) in Italy. Plant Dis 94(1):130–130
Tsay JG, Chen RS, Wang HL, Wang WL, Weng BC (2011) First report of powdery mildew caused by Erysiphe diffusa, Oidium neolycopersici, and Podosphaera xanthii on papaya in Taiwan. Plant Dis 95:1188
Acknowledgements
Funding for this work was provided by the National Key R&D Program of China (2018YFD0201100), the IAEA Collaborative Research Project D22005 (No. 20380), the International Exchange and Cooperation Project funded by the Agricultural Ministry ‘Construction of Tropical Agriculture Foreign Cooperation Test Station and Training of Foreign Managers in Agricultural Going-Out Enterprises’ (SYZ2019-08) and the Central Public-interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (No. 1630042017021).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this chapter

Cite this chapter
Wu, W. et al. (2023). A PCR-Based Assay for Early Diagnosis of the Coffee Leaf Rust Pathogen Hemileia vastatrix. In: Ingelbrecht, I.L., Silva, M.d.C.L.d., Jankowicz-Cieslak, J. (eds) Mutation Breeding in Coffee with Special Reference to Leaf Rust. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-67273-0_18
Download citation
DOI: https://doi.org/10.1007/978-3-662-67273-0_18
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-67272-3
Online ISBN: 978-3-662-67273-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)