
Group B Streptococcus colonization induces Prevotella and Megasphaera abundance-featured vaginal microbiome compositional change in non-pregnant women
- Published
- Accepted
- Received
- Academic Editor
- Jennifer Stearns
- Subject Areas
- Microbiology, Epidemiology, Gynecology and Obstetrics, Pediatrics
- Keywords
- Vaginal microbiome, Group B Streptococcus, Microbial colonization, Disease transmission
- Copyright
- © 2019 Mu et al.
- Licence
- This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. For attribution, the original author(s), title, publication source (PeerJ) and either DOI or URL of the article must be cited.
- Cite this article
- 2019. Group B Streptococcus colonization induces Prevotella and Megasphaera abundance-featured vaginal microbiome compositional change in non-pregnant women. PeerJ 7:e7474 https://doi.org/10.7717/peerj.7474
Abstract
Background
Previous studies have indicated that variations in the vaginal microbiome result in symptomatic conditions. Group B Streptococcus (GBS) is a significant neonatal pathogen and maternal vaginal colonization has been recognized as an important risk factor for neonatal disease. Therefore, it is important to discover the relationship between the composition of the vaginal microbiome and GBS colonization. This study explores the potential relationship between the composition of the vaginal microbiome and GBS colonization in non-pregnant Chinese women.
Methods
A total of 22 GBS-positive, non-pregnant women and 44 matched GBS-negative women were recruited for the current study. The composition of the vaginal microbiome was profiled by sequencing the 16S rRNA genes. The microbiome diversity and variation were then evaluated.
Results
The vaginal microbiome of the 66 subjects enrolled in the current study were compared and the results showed that GBS-positive women exhibited significant vaginal microbial differences compared with the GBS-negative women based on the analysis of similarities (r = 0.306, p < 0.01). The relative abundance of the bacterial genus Lactobacillus (p < 0.01) was significantly lower in the GBS-positive group, while the abundances of the bacterial genera Prevotella (p < 0.01), Megasphaera (p < 0.01), and Streptococcus (p < 0.01) were significantly higher in the GBS-positive group.
Discussion
The current study addressed significant variations across the communities of the vaginal microbiome in GBS-positive and GBS-negative women in a Chinese cohort, which paves the way for a larger cohort-based clinical validation study and the development of therapeutic probiotics in the future.
Introduction
Group B Streptococcus (GBS) is a Gram-positive bacteria that asymptomatically and transiently colonizes the gastrointestinal and vaginal tracts of healthy women. It is the leading cause of invasive bacterial disease in infants and can lead to a fatal infection in newborns (Korir, Manning & Davies, 2017). There are more than three million neonatal deaths worldwide each year, 20% of which are caused by infections, which is a global concern (Rick et al., 2017). Since the 1970s, GBS has been considered to be the main cause of perinatal infection. GBS colonization in infants less than 7-days old can cause septicemia and pneumonia, while in infants 7 days to 3 months of age, bacteremia and meningitis are often the major clinical complications of GBS colonization (Zaleznik et al., 2000). GBS is thought to contribute to chorioamnionitis, endometritis, urinary tract infections, sepsis, and premature birth in peripartum women (Krohn, Hillier & Baker, 1999; Yancey et al., 1994).
Since GBS is the leading cause of neonatal morbidity and mortality, several rapid tests based on molecular techniques and therapies for GBS have been developed and applied clinically (Davies et al., 2004). Pregnant women need to be screened between 34 weeks and 37 weeks of pregnancy and GBS-positive pregnant women should be considered for antibiotic treatment to protect newborns from GBS infection. Many antibiotics are widely believed to have an effect on newborns (Puopolo, Madoff & Eichenwald, 2005), therefore, probiotic therapy is currently considered to be an important adjuvant during antibiotic treatment. The proper long-term use of probiotics has not been found to negatively affect non-pregnant women and fetuses (Lundelin et al., 2017). The development of probiotic intervention strategies in the era of precision medicine relies on extensive research of the vaginal microbiome.
Previous studies have mainly examined the characteristics of GBS colonization in pregnant women. Vaginal bacterial cultures were taken from 4,025 women at 22 to 36 weeks of gestation and a significantly lower likelihood of coagulase-negative Staphylococcus, Prevotella, and Lactobacillus was found in the GBS-positive group (Kubota, Nojima & Itoh, 2002). It was observed that colonization by GBS could change the vaginal microbiome. GBS detection in healthy pregnant women correlated with a decrease in the Lactobacillus species (Altoparlak, Kadanali & Kadanali, 2004). In another study, there were no significant differences in the Lactobacillus or Bifidobacterium numbers based on GBS colonization (Brzychczy-Wloch et al., 2014). Recently, a study of 428 women in the USA showed that alpha diversity was not related to GBS status (Rosen et al., 2017). The discrepancy in the quantity of GBS-related taxa mentioned in these studies is probably due to different sampling locations, patient demographics, and sequencing strategies. Additionally, the relationship between GBS colonization and the vaginal microbiota can be affected by complex factors including education, ethnicity, geography, sexual behavior, and socioeconomic class (Albert et al., 2015). The previous studies mainly used culture-based methods to examine the relationship between GBS colonization and other components of the vaginal bacteria (Aveyard et al., 2002). However, this method often shows bacterial phenotypic expression instability and does not comprehensively reveal the relationship between GBS colonization and the microbiome in the female reproductive tract. The majority of the previous studies were also aimed at pregnant women (Kubota, Nojima & Itoh, 2002; Altoparlak, Kadanali & Kadanali, 2004). There were relatively few studies on non-pregnant women and especially few studies on the Asian cohort, which made GBS invasive bacterial disease in infants difficult to determine without adequate references. Therefore, we performed a pilot study to evaluate the potential relationship of the composition of the vaginal microbiome with GBS colonization in non-pregnant Chinese women.
Materials & Methods
Participants and study design
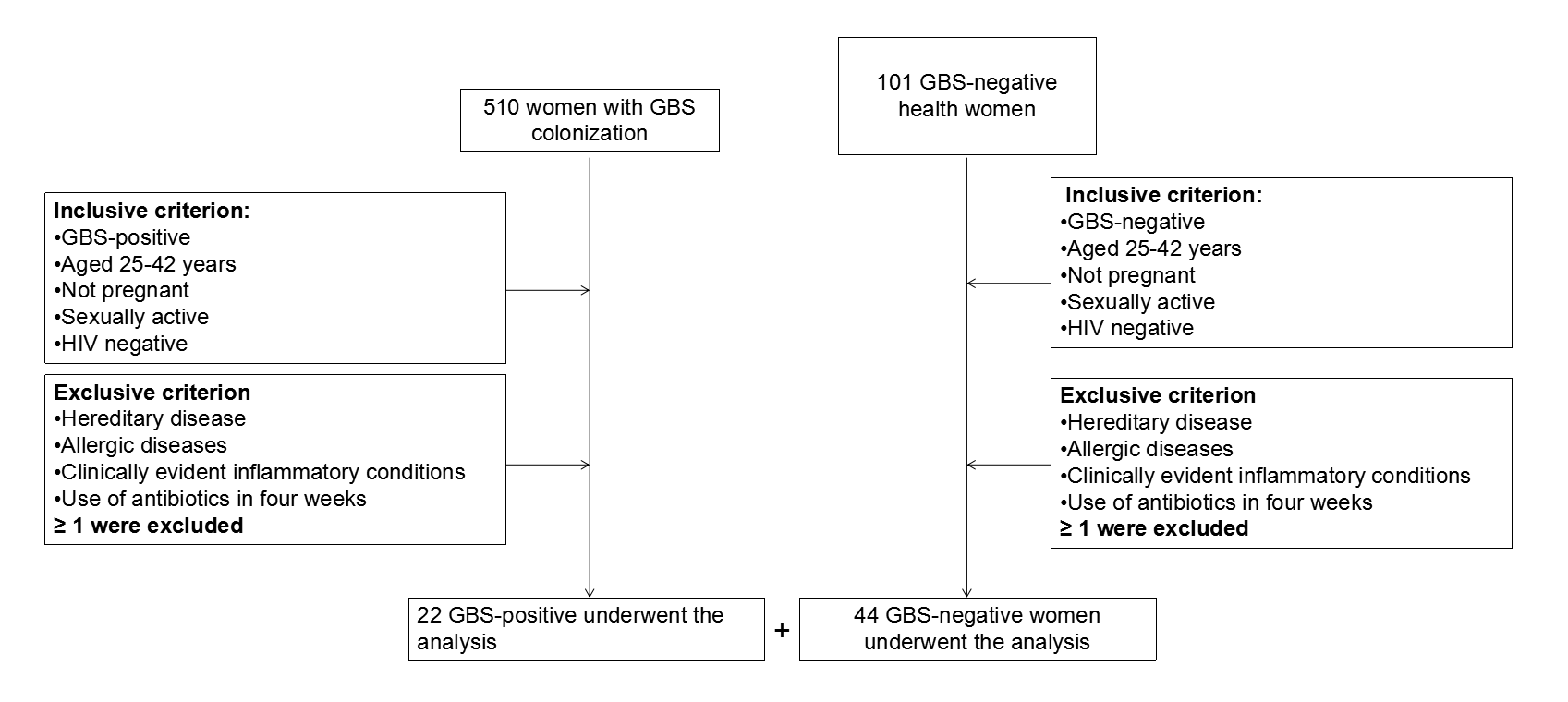
The current study was carried out in accordance with the recommendations of the Human Specimen Study Guidelines of the Institutional Review Board of the Affiliated Central Hospital of Qingdao University (IRB# QCH17-0328-01). Written informed consents were obtained from all patients before they were randomly assigned to groups. This research was conducted in compliance with national legislation and the Code of Ethical Principles for Medical Research Involving Human Subjects from the World Medical Association (Declaration of Helsinki). Women aged 25–42 years seeking primary gynecologic care were enrolled from March 2017 to December 2017 at the Qingdao Medical Center (Table 1). Over 600 samples were collected from individuals with GBS colonization and healthy individuals and vaginal samples for the current microbiome study were chosen from this specimen bank. The inclusion criteria for the current study were: (a) Women between the ages of 25–42 years, (b) non-pregnant, (c) sexually active, (d) HIV negative, (e) without clinically evident inflammatory conditions, and (f) women who didn’t use any oral or intravaginal antibiotic or antifungal medications for three months prior to the sample collection (Figure S1).
| GBS-P (n = 22) | GBS-N (n = 44) | P-value | |
|---|---|---|---|
| Age (year) | 29.86 ± 3.76 | 30.15 ± 4.02 | 0.7662 |
| Parity | 0.8153 | ||
| primiparity | 18 (81.82%) | 37 (84.10%) | |
| multiparity | 4 (18.18%) | 7 (15.91%) | |
| Use of hormonal contraceptives | 2(9.10%) | 3(6.82%) | 0.8694 |
| History of bacterial vaginitis | 5(22.73%) | 8(18.18%) | 0.6616 |
| Vaginal cleanliness | 0.5873 | ||
| I–II grade | 13(59.09%) | 29(65.9%) | |
| III–IV grade | 9(40.91%) | 15(34.1%) |
A GBS nucleic acid detection kit (BioChain, Beijing, China) was used to conduct preliminary testing for GBS colonization. Out of a total of 66 women screened, 22 women were GBS-positive and the other 44 were GBS-negative. Vaginal swab samples were collected, processed, and transferred to −80 °C storage within 30 min. In order to prevent contamination, strict standards and procedures were adopted. Sexual intercourse, tub bath, vaginal examination, vaginal lavage, and localized medication were prohibited 24 h before the collection of the vaginal specimens in order to avoid affecting the results of the examination. Sterilized scrapers, straws, or cotton swabs used for collecting samples were clean and dry without any chemicals or lubricants. The vaginal speculum could be moistened with a little saline, if necessary, before insertion. The swabs were placed directly into the sterile container.
Microbial DNA extraction, 16S rRNA gene amplicon sequencing
DNA was isolated from the vaginal swabs using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Valencia, CA, USA) (Sundquist et al., 2007). The DNA concentration was measured by NanoDrop ND-2000 (Thermo Fisher Scientific, Waltham, MA, USA). The V1–V2 hyper-variable region of the bacterial 16S rRNA gene was identified to analyze the microbial community within the samples. Two universal bacterial 16S rRNA gene amplicon PCR primers (PAGE purified) were used: forward primer-27F (5′AGAGTTTGATCMTGGCTCAG3′) and reverse primer-355R (5′GCTGCCTCCCGTAGGAGT3′). Amplicons were then purified using the QIAquick PCR Purification Kit (Qiagen) PCR purification procedure, quantified using a NanoDrop ND-1000 Spectrophotometer (Nucliber, Madrid, Spain), and then pooled in equal concentrations. All amplicons were quantified and pooled to equalize concentrations for sequencing using HiSeq 2500 (Illumina, San Diego, CA, USA).
Sequence processing and statistical analysis
The 16S rRNA gene sequence paired-end data set was merged and filtered using the FLASH method described by Magoč and Salzberg (Magoc & Salzberg, 2011). All sequence analysis was conducted by the Quantitative Insights into Microbial Ecology (QIIME, version 1.9.1) software suite (Caporaso et al., 2010), as per the QIIME tutorial (http://qiime.org/). Chimeric sequences were removed using usearch61 (Edgar, 2010) with denovo models. Sequences were clustered against the 2013 Greengenes (13_8 release) ribosomal database’s 97% reference data set (http://greengenes.secondgenome.com/?prefix=downloads/greengenes_database/).
Sequences that did not match any entries in this reference were subsequently clustered into de novo OTUs at 97% similarity with UCLUST. Taxonomy was assigned to all OTUs using the RDP classifier (Cole et al., 2009) within QIIME and the Greengenes reference data set.
OTU tables were subsequently subjected to abundance-based filtering, removing low-abundance OTUs that represented less than 0.005% of total reads in the data set. The OTU table was then rarefied to a sequencing depth of 25000 per sample, for subsequent analyses of alpha and beta diversity. Alpha diversity (observed OTUs and Shannon) and beta diversity (PCoA analysis) were analyzed using the QIIME standard pipeline.
Statistical analysis
The data were presented as the mean ± SD. The chi-squared-test was used to assess gender differences, and the Kolmogorov–Smirnov test was used to evaluate alpha diversity. An analysis of similarities (ANOSIM) on beta diversity matrices was conducted in QIIME to test for significant differences between the microbial communities. The significance of the ANOSIM test was assessed with 999 permutations. Linear discriminant analysis effect size (LEfSe) was introduced to identify bacterial biomarkers for two groups and was performed on the web-based Galaxy interface (http://huttenhower.sph.harvard.edu/galaxy) (Segata et al., 2011). Standard parameters were used, except for the alpha value for the factorial Kruskal-Wallis test among classes (alpha < 0.01) and the threshold used to consider a discriminative feature for the logarithmic LDA score was set at >3. Statistical dependence between continuous variables was determined using a Spearman’s rank correlation.
Results
Vaginal swab samples were collected from 66 female patients who met the inclusion criteria, which included 22 GBS-positive women and 44 GBS-negative women. The clinical characteristics of the subjects are summarized in Table 1. The age of the subjects ranged from 25 to 42 years, with an average age of 31.40 ± 4.05 years (GBS-negative group, 32.15 ± 4.02; GBS-positive group, 29.86 ± 3.76).
Different composition of vaginal microbiome in GBS-positive female patients
An analysis of alpha diversity revealed that there was no significant differences in the Shannon index between the GBS-negative and GBS-positive groups. (Fig. 1, Wilcoxon rank sum test, p = 0.2). Beta diversity, based on the unweighted and weighted UniFrac distance, was significantly different between the two groups (r = 0.23, p = 0.001, unweighted Unifrac Distance, Fig. 2, ANOSIM).
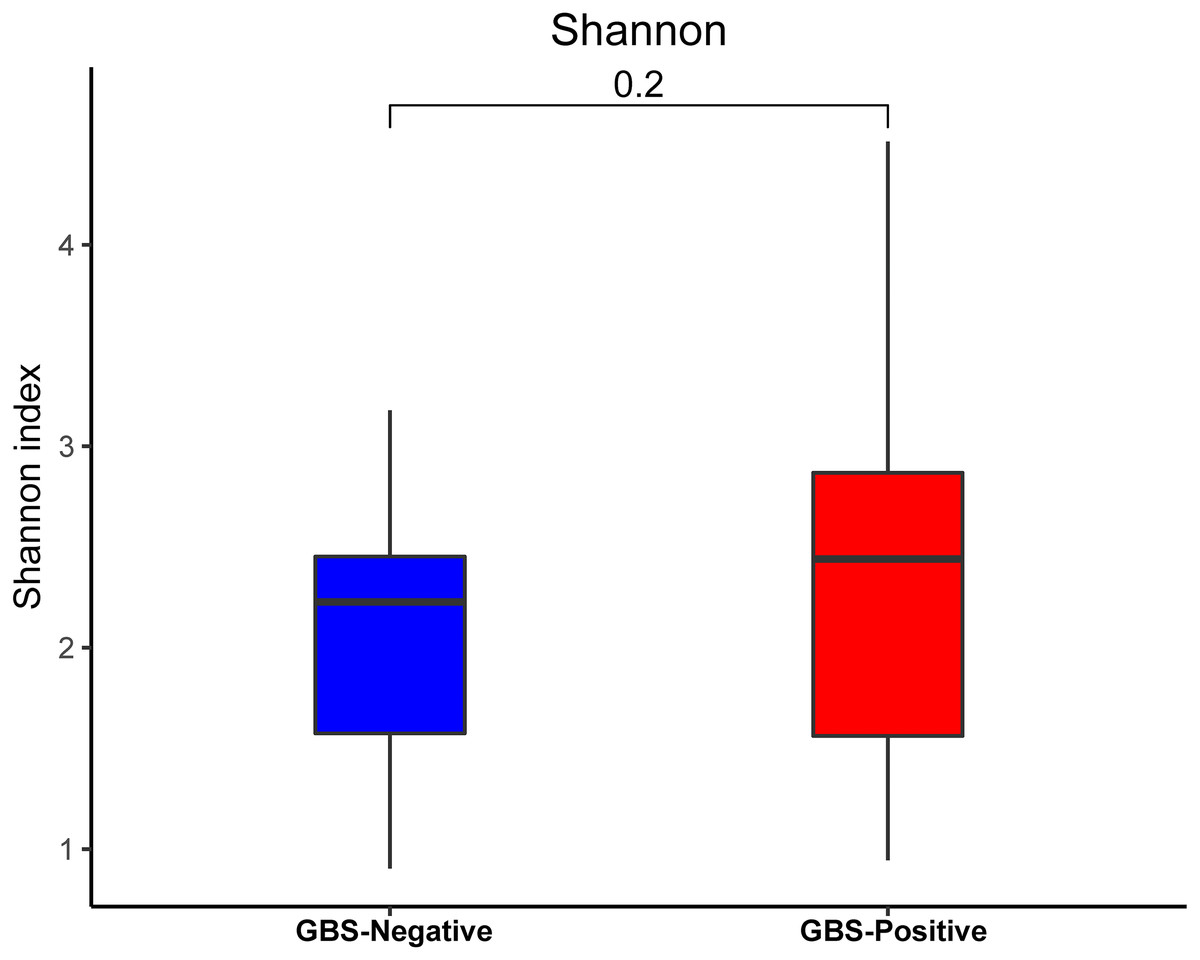
Figure 1: Comparison of the α-diversity (Shannon index) based on the OTUs profile in GBS-positive group and GBS-negative group.
The p value was calculated by the Wilcoxon rank-sum test.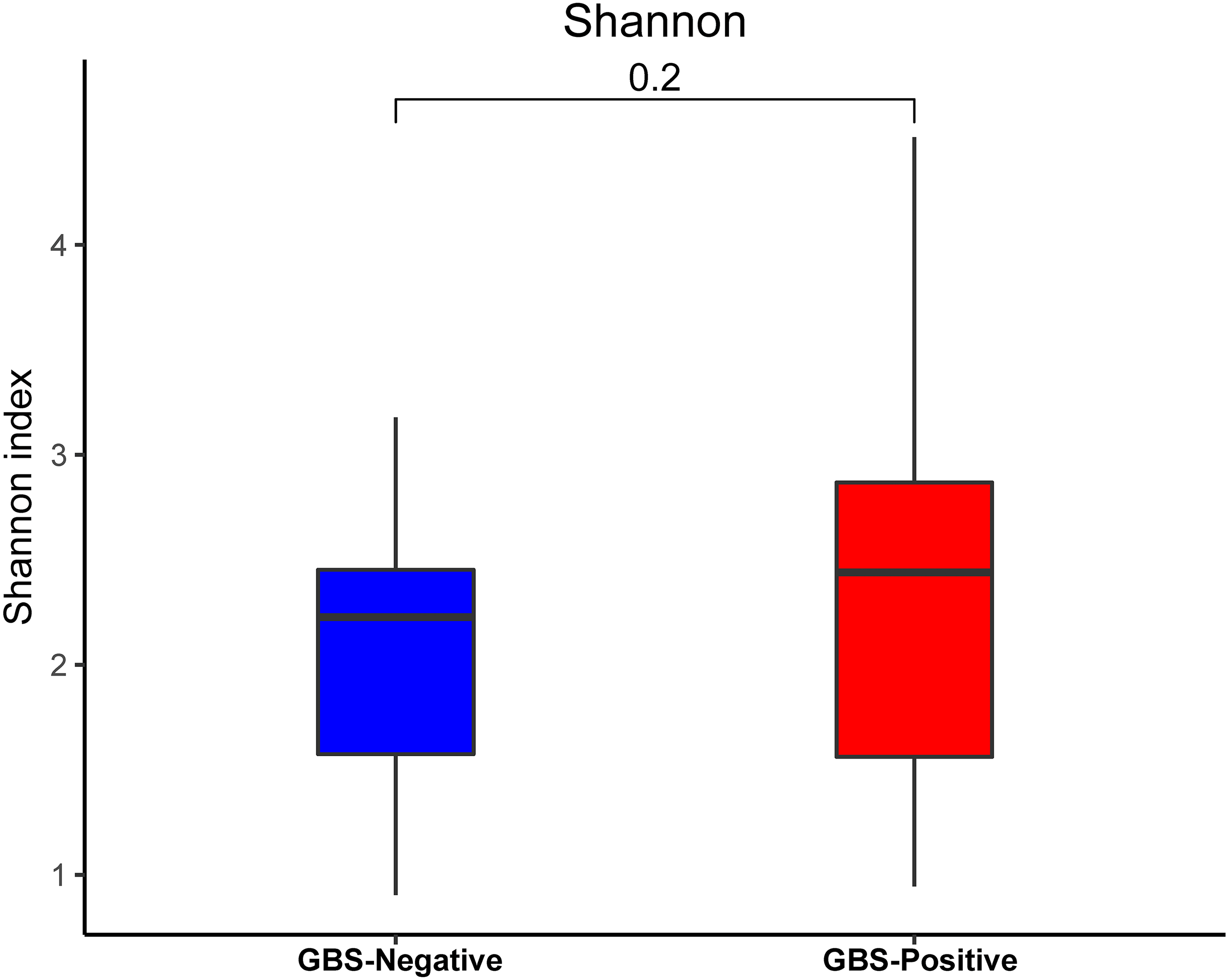{kind=link}
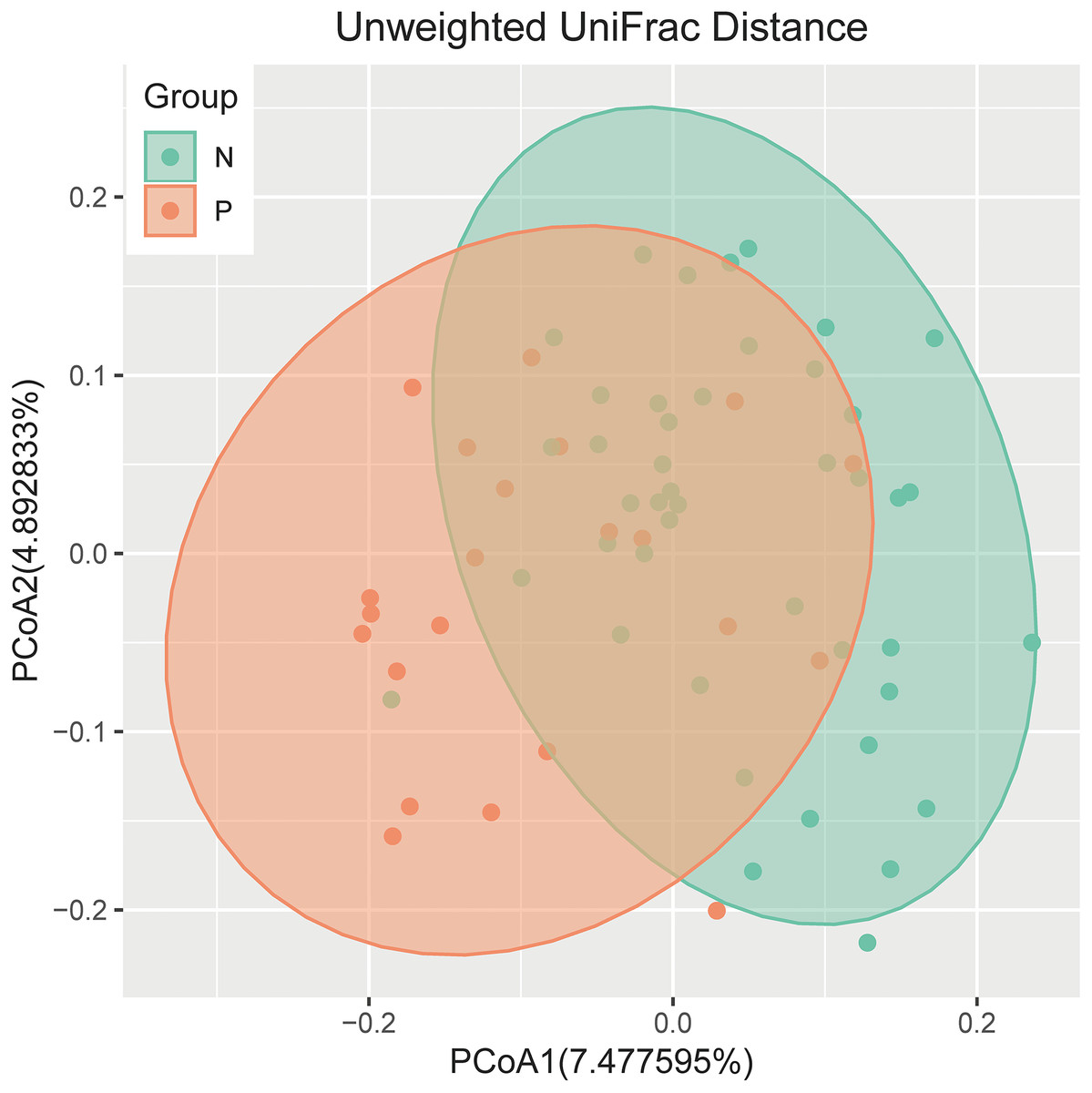
Figure 2: PCoA of bacterial beta diversity based on the unweighted UniFrac distance and weighted UniFrac distance.
GBS-negative and GBS-positive groups are colored in green and orange, respectively.{kind=link}
Relative taxon abundance in the vaginal microbiome of patients with GBS-positive and GBS-negative groups
Using 97% as the similarity cut off, 356 qualified taxa were identified. The microbiome profiles in the vaginal samples of patients were dominated by the phylum Firmicutes (90.6% ± 16.3%), Actinobacteria (5.1% ± 13.8%), Bacteroidetes (2.6% ± 7.1%), Proteobacteria (7.5% ± 1.1%), and Tenericutes (0.6% ± 1.5%). A phylum level analysis also demonstrated that the number of Firmicutes was less abundant in patients with GBS (Fig. 3A). At the genus level, the abundances of Prevotella, Megasphaera, and Streptococcus were significantly higher, while Lactobacillus was slightly lower in the GBS-positive group compared to the GBS-negative group (Fig. 3B).
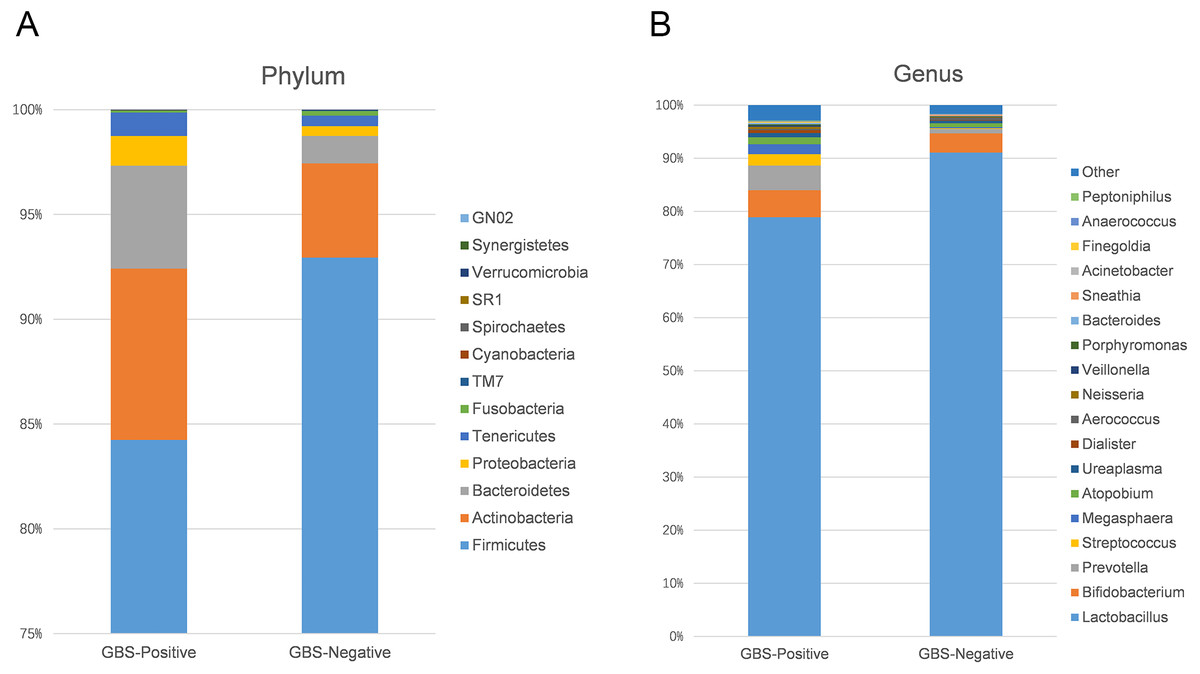
Figure 3: Comparison of relative taxa abundance between GBS-negative and GBS-positive groups at the phylum (A) and genus levels (B).
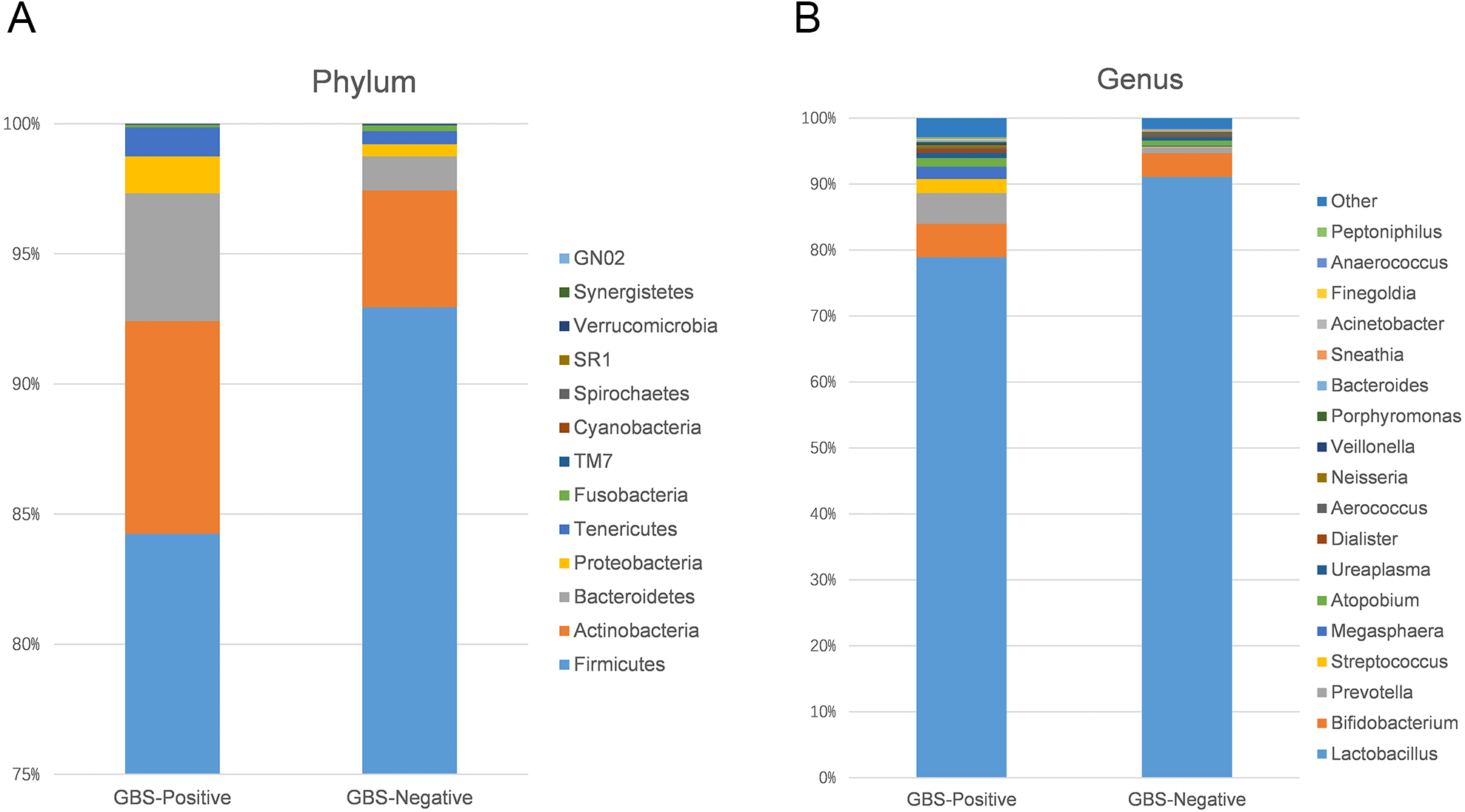{kind=link}
Differential taxonomic abundance in the vaginal microbiome of patients with GBS-positive and GBS-negative groups
A total of 54 taxa were found to be significantly associated with GBS, using LEfSe analysis (logarithmic discriminant analysis score >2.0). Only 15 taxa remained after filtering for those with an absolute value of the logarithmic discriminant analysis score ≥3.0 (Fig. 4). LEfSe analysis revealed a significantly higher relative abundance of some bacterial taxa in the GBS-positive group when compared with those in the GBS-negative group. These included Bacteroidetes (phylum), Bacteroidia, Clostridia (class), Bacteroidales, Clostridiales (order), Prevotellaceae, Veillonellaceae, Streptococcaceae (family), Prevotella, Megasphaera, and Streptococcus (genus), with a significantly lower relative abundance of taxa Firmicutes (phylum), Bacilli (class), Lactobacillales (order), Lactobacillaceae (family), Lactobacillus (genus) in GBS-positive vs. GBS-negative women (p <0.01, Wilcoxon rank-sum test; LDA >3.0).
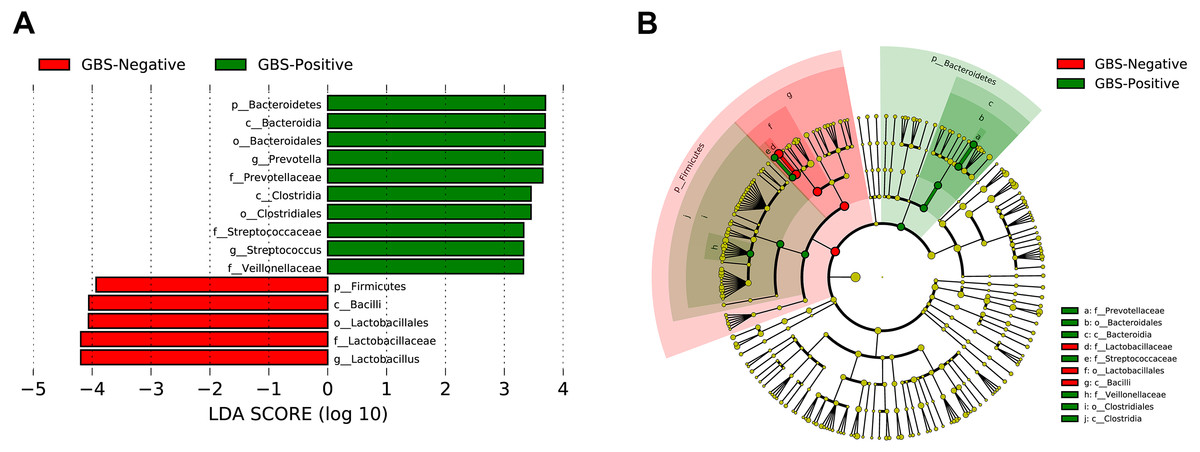
Figure 4: Characteristics of microbial community composition in GBS-negative and GBS-positive groups.
(A) The most differentially abundant taxa between GBS-negative and GBS-positive groups (LDA score above 3) which was generated from LEfSe analysis; (B) The enriched taxa in GBS-negative and GBS-positive groups fecal microbiota were represented in Cladogram. The central point represents the root of the tree (Bacteria), and each ring represents the next lower taxonomic level (phylum to genus: p, phylum; c, class; o, order; f, family; g, genus). The diameter of each circle represents the relative abundance of the taxon.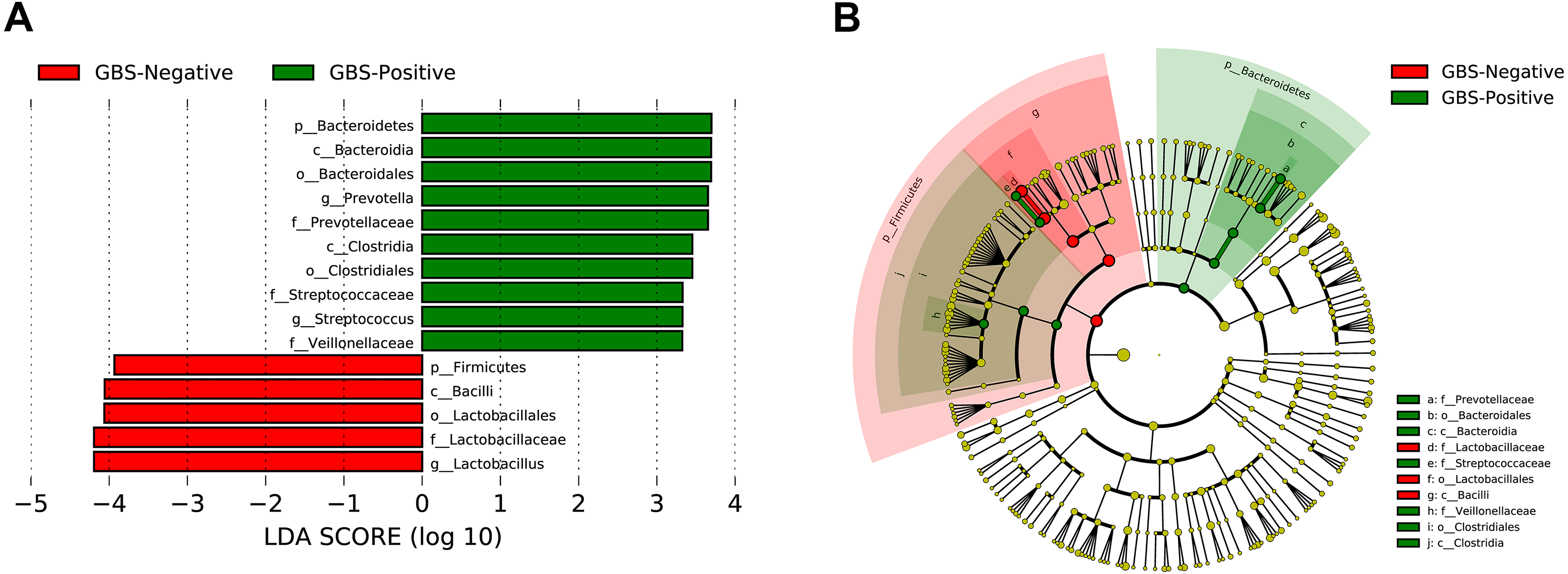{kind=link}
Correlation of candidate bacterial taxa
The taxa that met the threshold of significance after modeling were considered candidate bacterial taxa. Correlation between each of these candidate bacterial taxa was examined (Fig. 5). There were statistically significant positive correlations between each of the candidate taxa associated with GBS-negative status. There were statistically significant positive correlations between the genus Porphyromonas and Prevotella and the genus Streptococcus (p < 0.01). Lactobacillus showed significant negative correlations with the genus Streptococcus.
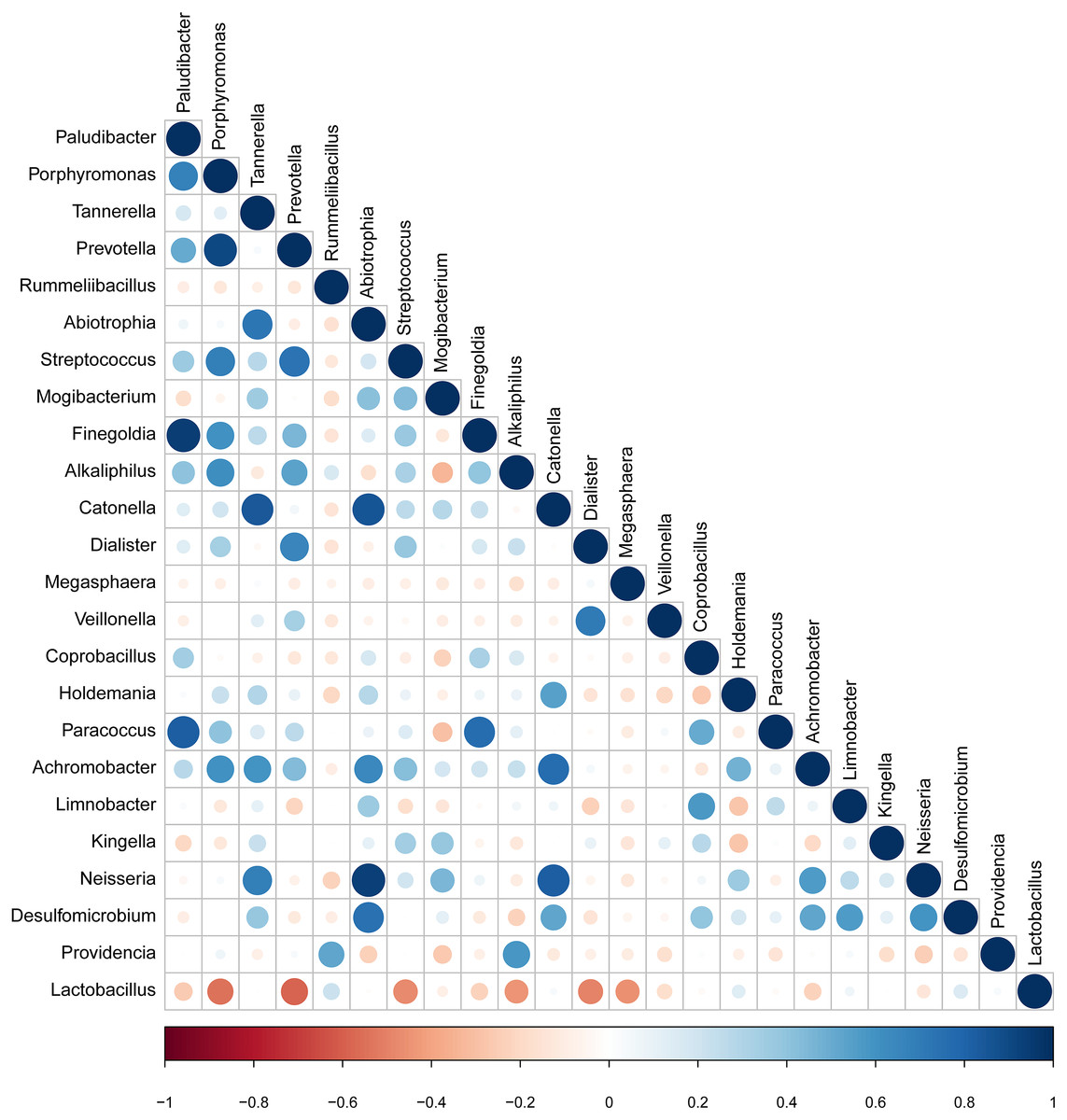
Figure 5: Correlation between candidate taxa. (The genus of significant difference between GBS positive and GBS negative was selected ( p < 0.05, LDA > 2)).
There were statistically significant positive correlations between the genus Porphyromonas and Prevotella and the genus Streptococcus. While Lactobacillus showed significant negative correlations with the genus Streptococcus.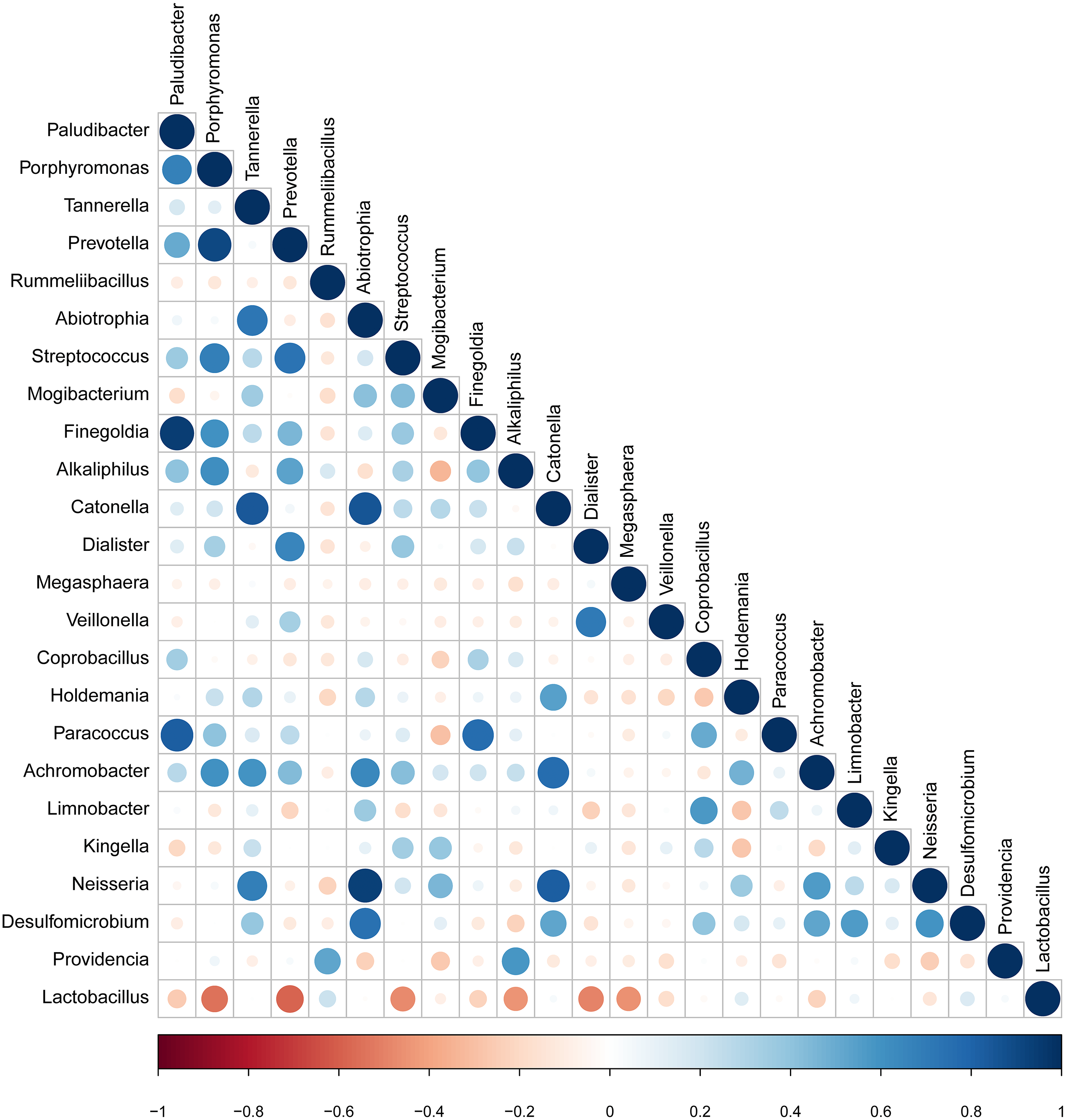{kind=link}
Discussion
It is well-recognized that maternal vaginal colonization with GBS is a major risk factor for the early onset of invasive GBS disease in newborns (Centers for Disease Control and Prevention, 1996). However, the relationship between GBS and the vaginal microbiome is relatively less well-known in the Chinese cohort. The current study aimed to determine the influence of GBS colonization on Chinese women. Additionally, as an exploratory objective, and given the increasing evidence suggesting that the microbiome is an important determinant of vaginal pathogen colonization (Balkus et al., 2017; Datcu et al., 2013; Cruciani et al., 2015), the potential relationship between the composition of the vaginal microbiome and GBS colonization was analyzed in this population. It was confirmed that there is a unique microbiome in the GBS-positive group compared with the GBS-negative group. Also, the microbiome profiles of GBS-negative and GBS-positive groups were compared and microbial biomarkers were identified.
Many studies have shown that the normal vaginal microbiome is largely characterized by the dominance of a single OTU, most closely related to the Lactobacillus species (Ahmed et al., 2012; Albert et al., 2015; Paramel Jayaprakash, Schellenberg & Hill, 2012). The Lactobacillus species maintain an acidic pH in the vagina and inhibit pathogenic microorganisms (Bhandari & Prabha, 2015). The majority of healthy vaginal microbiota are dominated by Lactobacillus. In the current study, GBS colonization was negatively correlated with the abundance of Lactobacillus. An earlier study reported that there was no relationship between GBS colonization and the vaginal microbiome alpha diversity (Rosen et al., 2017). We were consistently unable to find a significant difference in the alpha diversity within the microbiome of GBS-positive and negative groups.
Individual bacterial taxa which have associations with bacterial vaginosis have had mixed associations with GBS colonization (Discacciati et al., 2011). In this study, OTUs from the genera Prevotella, Megasphaera, and Streptococcus, and OTUs from the families Prevotellaceae, Veillonellaceae, and Streptococcaceae were found to be more abundant in the GBS-positive group compared with the GBS-negative group. Prevotella, commonly identified in the vagina, is associated with bacterial vaginosis (BV), and has been associated with GBS-positive status (Datcu, 2014; Dols et al., 2011; Ling et al., 2010). Megasphaera is also associated with a GBS-positive status. Its presence can be highly correlated with BV (Datcu, 2014; Dols et al., 2011; Balkus et al., 2017; Ling et al., 2010). These complex relationships, combined with alpha diversity analysis and other studies suggest that GBS colonization is related to BV and to the presence or absence of specific bacterial members of the vaginal microbiome. Additionally, a decrease in Lactobacillus in the GBS positive group was found (Figs. 3B and 4). It has been reported that Lactobacillus of vaginal origin can inhibit the attachment of genital uropathogenic GBS to the vaginal epithelium (Zarate & Nader-Macias, 2006). Lactobacillus has also shown a significant negative correlation with the abundance of the genus Streptococcus (Figurec5). Furthermore, the current study indicated a statistically significant and positive correlation between the genera Porphyromonas and Prevotella, and the genus Streptococcus. This finding suggests that further studies need to be performed to better understand the relationship between GBS colonization and the overall vaginal microbiome community structure.
Currently, studies on the microbiome support the application of intestinal probiotic supplements for healthy intestinal immune function and microbiome balance (Dimidi, Rossi & Whelan, 2017; Nishiyama et al., 2017; Seo et al., 2017). The vaginal microbiome remains less explored than the gut microbiome although it comprises a large proportion of the female microbial network (Albert et al., 2015; Power, Quaglieri & Schulkin, 2017). Preliminary studies and data from a large health maintenance organization showed that 40–50% of GBS-colonized multiparous women were unable to receive antibiotics at least 4 h before delivery due to the rapidity of their labor (Davies et al., 2001). Most research to date has focused on the potential of probiotics to prevent bacterial vaginosis and preterm labor. The effectiveness of probiotics as a surrogate or adjunctive therapy for intrapartum antibiotic prophylaxis in GBS colonized women has rarely been evaluated (Ho et al., 2016). The findings of the current study support the idea that increasing the abundance of beneficial bacteria, such as Lactobacillus, can reduce GBS colonization. However, there are several limitations to the current study; for instance, the sample size was small and all patients came from a single hospital and resided in the city of Qingdao, China. Therefore, the findings may not be able to be generalized across different regions and races.
Probiotic therapies are the key to maintaining vaginal health (Bhandari & Prabha, 2015; De Gregorio et al., 2015; Power, Quaglieri & Schulkin, 2017; Surendran Nair, Amalaradjou & Venkitanarayanan, 2017; Woodman, 2016). More importantly, this potential application of probiotics needs to be investigated further in future studies in order to reduce early-onset GBS colonization and the need for antibiotic treatment during labor.
Conclusions
In conclusion, the current study has provided preliminary data for an association between GBS colonization and the vaginal microbiome. The colonization of potential vaginal pathogens, such as Prevotella, Megasphaera, and Streptococcus, was significantly higher in the GBS-positive group. Thus, specific microbial taxa were associated with colonization of this significant human pathogen, highlighting a potential role for the microbiota in the promotion or inhibition of GBS colonization.
{kind=link}