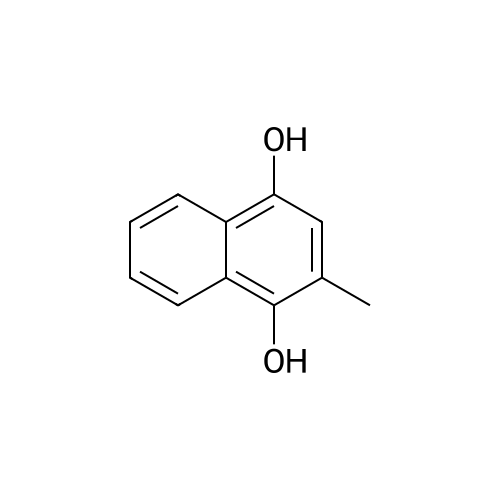
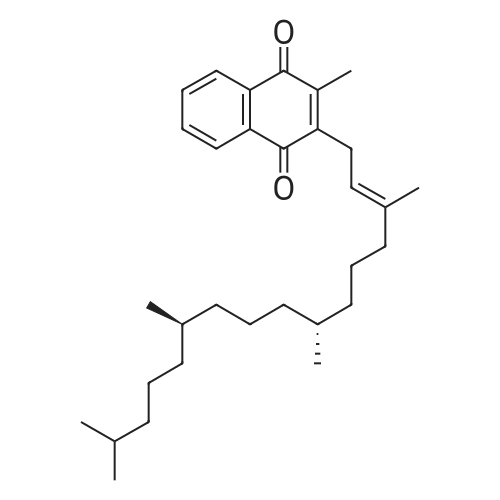
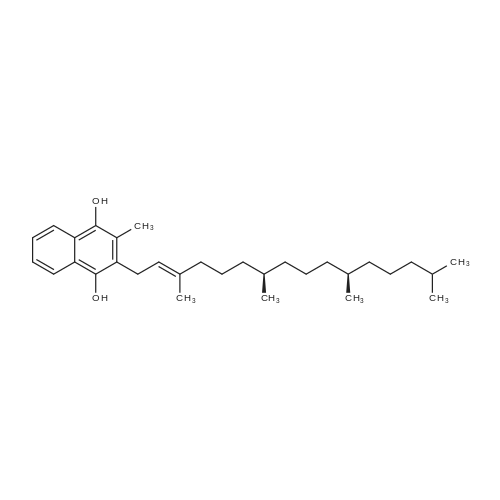
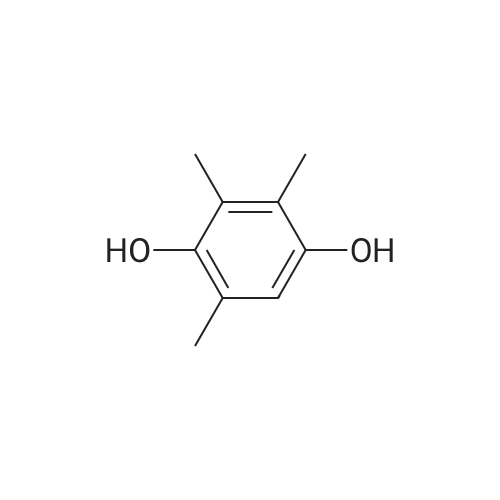
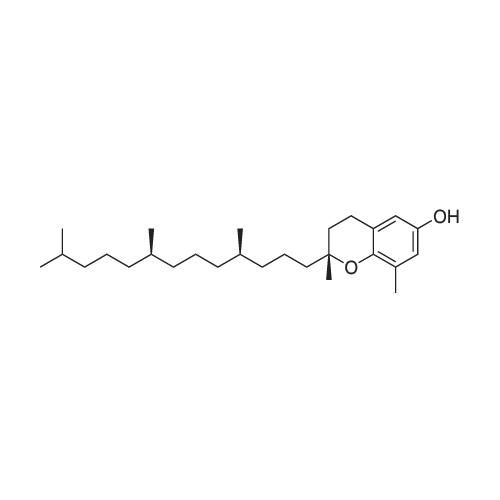
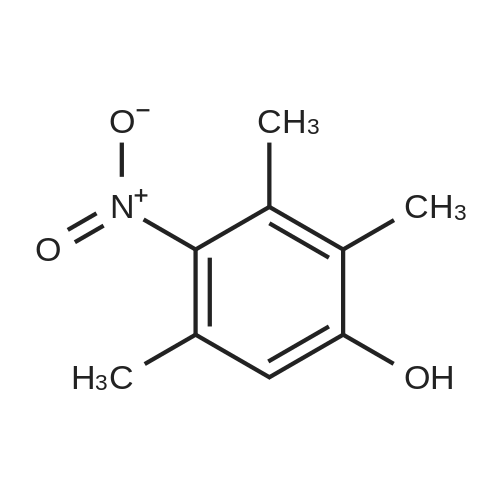
| 76% |
With triphenylphosphine; diethylazodicarboxylate In tetrahydrofuran; toluene at 0 - 20℃; for 2.1h; Inert atmosphere; |
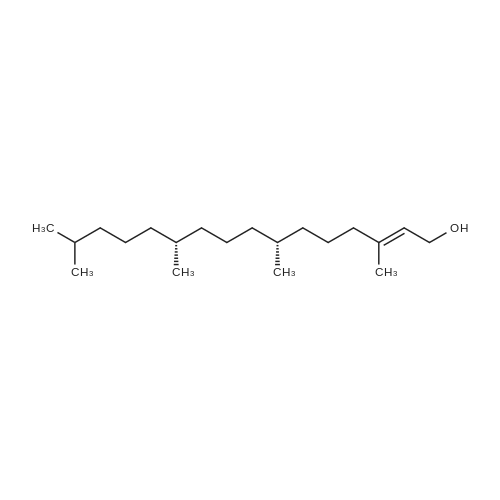
xxiv xxiv) 5-Fluoro-1-((3aR,4R,6R,6aR)-6-(((4-methoxyphenyl)diphenylmethoxy)-2,2-dipropyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)-3-((7R,11R)-3,7,11,15-tetramethylhexadec-2-en-1-yl)pyrimidin-2,4(1H,3H)-dione (4a (E+Z))
Compound 3 (1 g; 1.59 mmol) was dissolved in anhydr. THF (10.6 ml). After addition of phytol (0.61 ml; 1.59 mmol) and Ph3P (0.62 g; 2.38 mmol) , the reaction mixture was stirred for 5 min at room temp. under N2 atmosphere and with exclusion of light. Then, the reaction mixture was cooled to 0°C, and a 40% soln. of diethylazodicarboxylate (DEAD) in toluene (0.69 ml; 2.38 mmol) was added dropwise within 1 min. After further 5 min of stirring at 0°C the mixture was allowed to warm up to room temp., and stirring was continued for 2 h. After evaporation of the solvent in high vacuo (45°C) the residue was purified by repeated chromatography on silica gel (1. column: 2 x 25 cm, EtOAc/petrolether, 1:13; 2. column: 2 x 18 cm, EtOAc/petrolether, 15:75, each solvent with 1% of Et3N). Yield 1.1 g (76%) of a colourless foam. Rf (EtOAc/petrol ether, 1:7) 0.55/0.60 (E/Z isomers). UV (MeOH): 270 (9,000). 1H-NMR ((D6)DMSO): 8.16 (d, 3J( H-C(6)cis, F) = 6.5, H-C(6)cis)); 8.25 (d, 3J(H-C(6)trans, F) = 6.5, H-C(6)trans)); 7.38 - 7.21 (m, 12H, 2 x H-C(3"), 4 x H-C(8"), 4 x H-C(9"), 2 x H-C(10")); 6.84 (d, 2H, 3J(H-C(4"), H-C(3")) = 9.0, 2 x H-C(4")); 5.84 (s, H-C(1')); 4.99 - 4.97 (m, 2H, H-C(2'), H-C(2"')); 4.67 - 4.63 (m, H-C(3'); 4.36 (d, 3J(H-C(1"')cis, H-C(2"')) = 5.0, H-C(1'")cis); 4.33 (d, 3J(H-C(1"')trans, H-C(2"')) = 5.0, H-C(1"')trans); 4.21 - 4.18 (m, H-C(4')); 3.72 (s, 3H, H3-C(6")); 3.35 - 3.32 (m, Hα-C(5')); 3.15 - 3.09 (m, Hβ-C(5')); 2.07 (t, 2H, 3J(H-C(4"')cis, H-C(5"')) = 7.5, H-C(4"')cis); 1.87 (t, 2H, 3J(H-C(4"')trans, H-C(5"')) = 7.5, H-C(4"')trans); 1.67 (s, 3H, H3C(20"')); 1.66 - 1.63 (m, 2H, H2-C(α')); 1.51 - 1.43 (m, 3H, H2-C(α), H-C(15"')); 1.42 - 1.30 (m, 6H, H2-C(β'), H2-C(5"'), H-C(7"'), H-C(11")); 1.28 - 0.98 (m, 16H, H2-C(β), H2-C(6"'), H2-C(8"'), H2-C(9"'), H2-C(10"'), H2-C(12"'), H2-C(13"'), H2-C(14"')); 0.91 (t, 3H, 2J((Ha-C(γ'), Hb-C(γ')), ((Ha-C(γ'), Hc-C(γ'))) = -7.0, H3-C(γ')); 0.84 (t, 3H, 2J((Ha-C(γ), Hb-C(γ)), ((Ha-C(γ), Hc-C(γ))) = -7.0, H3-C(γ)).0.83 - 0.78 (m, 12H, H3-C(16"'), H3-C(17"'), H3-C(18"'), H3-C(19"')). 13C-NMR ((D6)DMSO): 158.15 (C(5")); 156.09 (d, 2J(C(4), F) = -25.78, C(4); 148.60 (d, 4J(C(2), F) = 6.28, C(2)); 144.03 (C(7")); 139.35 (d, 1J(C(5), F) = 229.76, C(5)); 139.84 (s, C(3"')cis; 139.56 (s, C(3"')trans); 134.67 (C(2")); 129.86 - 126.74 (m, C(3"), C(8"), C(9"), C(10")); 125.55 (d, 2J(C(6), F) = -32.82, C(6)); 117.83 (C(2"')); 116.71 (C(Ketal)); 113.05 (C(4")); 93.22 (C(4')); 86.33 (C(1")); 85.95 (C(1')); 83.74 (C(2')); 80.82 (C(3')); 64.13 (C(5')); 54.88 (C(6")); 39.00 (C(1"')); 38.66 (C(α')); 38.54 (C(α)); 36.65-36.51(m, C(6"'), C(8"'), C(10"'), C(12"')); 35.81, 35.70 (2s, C(7"'), C(11'")); 27.25 (C(15"')); 24.27 (C(5"')); 24.01 (C(9"')); 23.62 (C(13"')); 22.41, 22.32 (2s, C(16"'), C(17"')); 19.48, 19.43 (2s, C(18"'), C(19"')); 16.88 (C(β')); 16.27 (C(β)); 15.85 (C(20"')); 14.05 (C(γ')); 14.02 (C(γ)). Anal. calc for C56H77FN2O7 (909.218): C, 73.98; H, 8.54; N, 3.08. Found : C, 73.75; H, 8.57; N, 2.73. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping