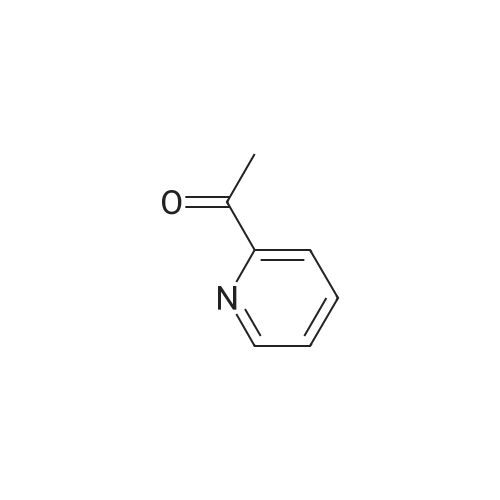
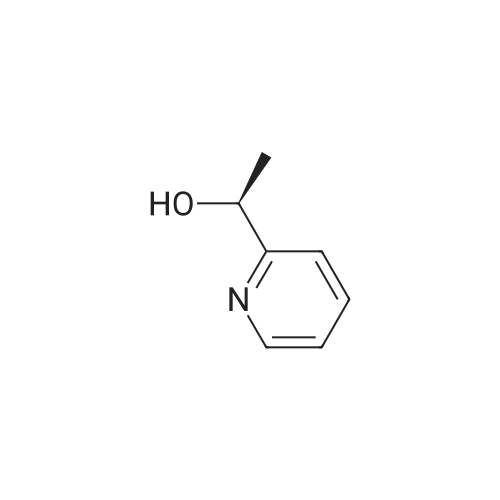
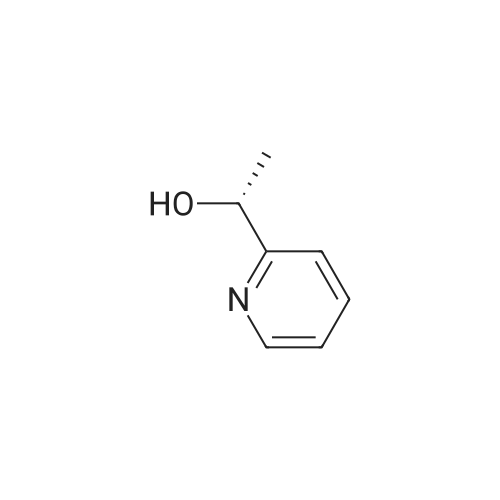
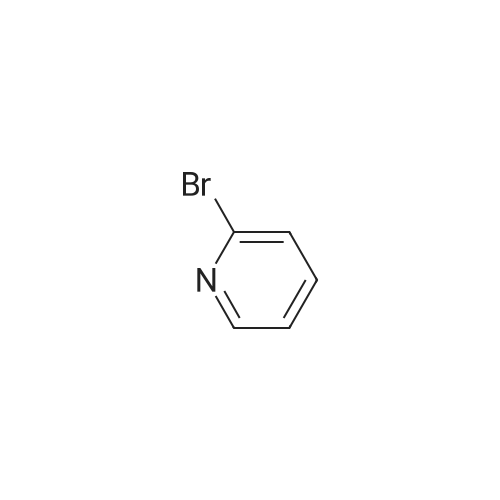
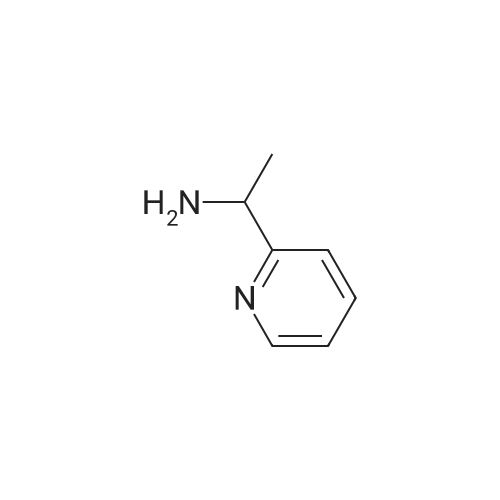
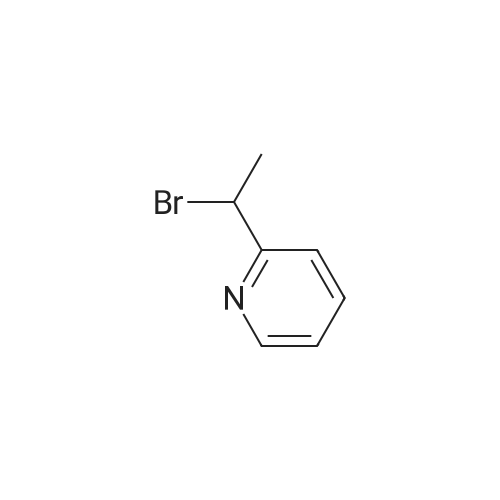
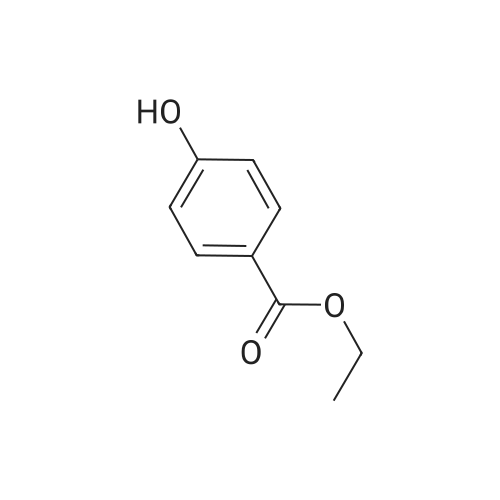
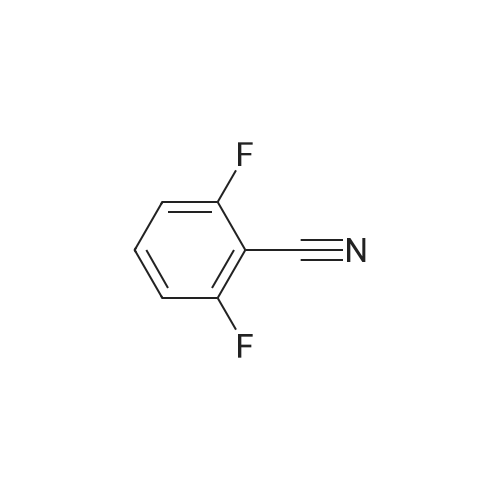
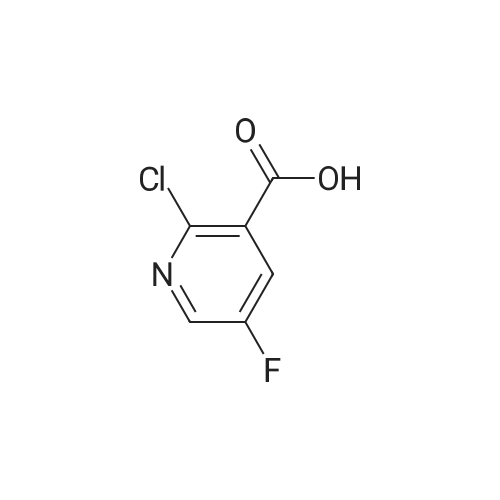
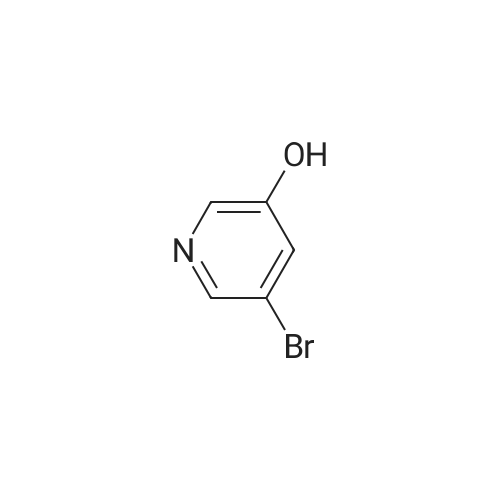
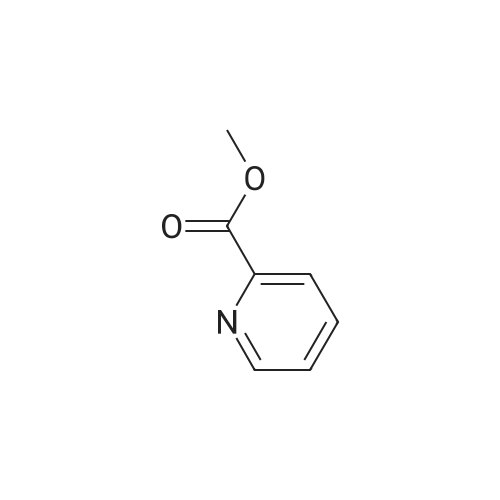
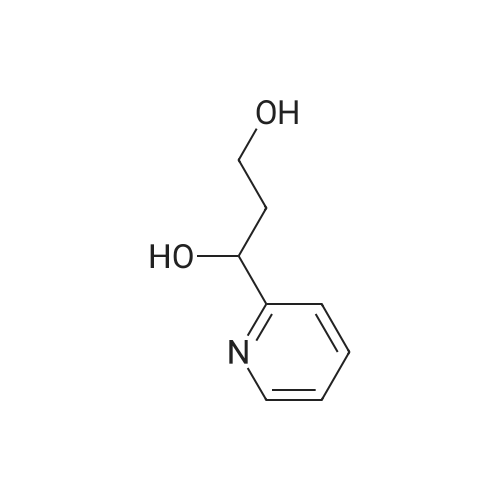
| 100% |
With [(CH2(NC(C6H5)N(C4H9)CHC)2)RhI2((C6H5)2PC2H4P(C6H5)2)](1+)*I(1-)=[(CH2(NC(C6H5)N(C4H9)CHC)2)RhI2((CH2P(C6H5)2)2)]I; isopropanol; potassium hydroxide In water monomer for 24h; Reflux; |
|
| 99% |
With stannous trifluoromethanesulfonate; PMHS; 2,6-bis(4,5-dihydrooxazol-2-yl)pyridine In methanol at 20℃; for 14h; |
|
| 99% |
With formic acid; triethylamine In ethyl acetate at 24℃; for 18h; |
|
| 99% |
With lithium isopropoxide In isopropanol at 180℃; for 0.333333h; microwave irradiation; |
|
| 99% |
With formic acid; triethylamine In ethyl acetate at 25℃; for 15h; Inert atmosphere; |
|
| 99% |
With [(iPr-PNP)Fe(H)(CO)(η1-BH4)]; hydrogen In ethanol at 40℃; for 6h; Inert atmosphere; |
|
| 99% |
With hydrogen In ethanol at 120℃; for 16h; Autoclave; |
|
| 99% |
With C25H30ClIrN2O3; water monomer; potassium hydroxide In isopropanol for 1h; Reflux; |
|
| 98% |
With potassium hydroxide; isopropanol for 10h; Heating; |
|
| 98% |
With formic acid; C18H24ClIrN3 In water monomer at 80℃; for 12h; Schlenk technique; Inert atmosphere; chemoselective reaction; |
|
| 97% |
With isopropanol; lithium tert-butylate at 180℃; for 0.5h; |
|
| 97% |
With trimethylamine-N-oxide; (N,N,N-trimethyl-2-(5-oxo-4,6-bis(trimethylsilyl)cyclopenta[c]pyrrol-2-(1H,3H,5H)-yl)ethanaminium) iron tricarbonyl; hydrogen In water monomer at 85℃; for 14h; Autoclave; Inert atmosphere; |
|
| 97% |
With [Re(NH{CH2CH2P(iPr2)}2)(CO)3]Br; potassium-t-butoxide; hydrogen In toluene at 110℃; for 17h; Inert atmosphere; Glovebox; Autoclave; |
|
| 96% |
With isopropanol at 85℃; for 24h; Inert atmosphere; |
|
| 96% |
With Cp*Ir(6,6'-dionato-2,2'-bipyridine)(H2O); hydrogen In tert-Amyl alcohol at 30℃; for 12h; Green chemistry; |
4.2. General procedure for catalytic hydrogenation of 2
General procedure: To an oven-dried 5 mL round-bottom flask were added ketone (1 mmol), cat. 7 (2.7 mg, 0.5 mol %) and tert-amyl alcohol (1 mL). Next, vacuum was applied to the flask followed by filling with H2 gas and keeping the flask attached to a balloon filled with H2 gas. The mixture was heated at 30 °C for 12 h. After completion of the reaction, the solvent was removed by evaporation under reduced pressure. The alcohols were isolated and purified by filtering a hexanes/ethyl acetate (5:1) solution of the crude product through a pad of silica gel, and then removing the solvent under reduced pressure. The conversion and purity of the alcohol products was assessed using NMR spectroscopy. |
| 96% |
With methanol; [Cp*Ir(2,2'-bpyO)(OH)][Na] at 66℃; for 12h; Inert atmosphere; Schlenk technique; |
|
| 95% |
With sodium tetrahydridoborate; poly[N-(2-aminoethyl)acrylamido]trimethyl ammonium chloride In tetrahydrofuran; water monomer at 20℃; for 0.75h; |
|
| 93% |
With benzyltriphenylphosphonium tetraborate In methanol at 20℃; for 0.0833333h; |
|
| 93% |
With [(N,N′-bis(diisopropylphosphino)-2,6-diaminopyridine)Mn(CO)3][Br]; potassium-t-butoxide; hydrogen In toluene at 130℃; for 38h; Glovebox; Autoclave; |
2.2. Typical catalytic hydrogenation
General procedure: In a glove box, an autoclave was charged with the desired ketone (0.5 mmol), toluene (2 mL), Mn complex 1 (14 mg, 5 mol%) followed by t-BuOK (5.6 mg, 10 mol%), in this order. The autoclave is then closed and charged with H2 (50 bar). |
| 93% |
With Cp*Ir(6,6'-dionato-2,2'-bipyridine)(H2O); isopropanol at 82℃; for 6h; Inert atmosphere; Green chemistry; |
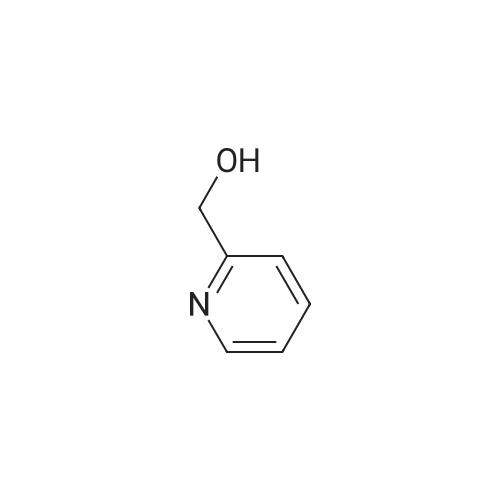
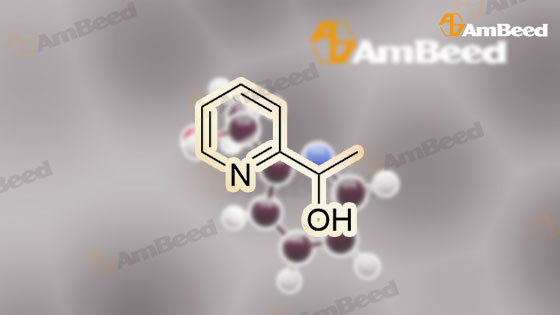
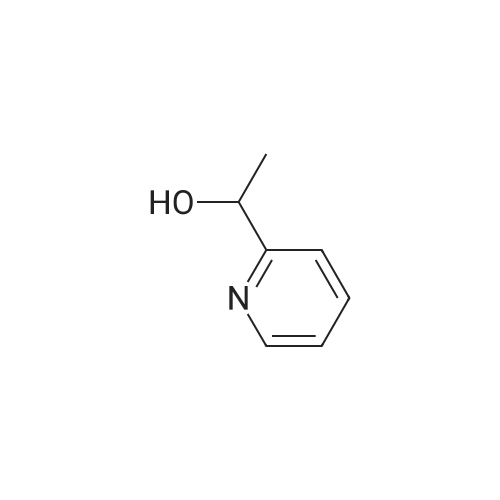
15 1-(Pyridin-2-yl)ethanol
The 2 - acetyl pyridine (121 mg, 1.0 mmol), cat. [Ir] (1.1 mg, 0 . 002 mmol, 0.2 μM %) and isopropyl alcohol (5 ml) are added to the 25 ml Kjeldahl tube, N2Protection Cooling to room temperature, rotary evaporation to remove the solvent, then through the column chromatography (developing solvent: petroleum ether/ethyl acetate) to obtain the pure target compound, yield: 93% |
| 93% |
With [Cp*Ir(2,2'-bpyO)(OH)][Na]; hydrogen In water monomer at 30℃; for 12h; Green chemistry; |
4.1. General procedure for catalytic hydrogenation of ketones,aldehydes or unsaturated aldehydes
General procedure: To an oven-dried 5 mL round-bottom flask were added ketonesor aldehydes or unsaturated aldehydes (1 mmol), cat. 6 (5.5 mg,1 mol %) and H2O (1 mL). Next, vacuum was applied to the flask followedby filling with H2 gas and keeping the flask attached to a balloonfilled with H2 gas. The mixture was heated at 30 °C for 12 h.After completion of the reaction, the mixture was extracted withethyl acetate (5 mL x 3). Then, the ethyl acetate layers were combined, dried with anhydrous sodium sulfate, filtered, and concentratedby evaporation under reduced pressure. The alcohols wereisolated and purified by filtering a hexanes/ethyl acetate (8:1)solution of the crude product through a pad of silica gel. Thenthe solvent was removed under reduced pressure to afford the correspondingproducts. The purity of alcohol products was assessedusing 1H NMR spectroscopy. |
| 93% |
With [Cp*Ir(2,2'-bpyO)(OH)][Na]; hydrogen In water monomer at 30℃; for 12h; |
17 The method is:
2-Acetylpyridine (121 mg, 1.0 mmol), metal ruthenium complex [Cp*Ir(2,2'-bpyO)(OH)][Na] (4.6 mg, 0.01 mmol, 1 mol %) and water (1 mL) sequentially added to 25 ml round bottom flasks, and replace the air in the round bottom flask with hydrogen. The pressure of hydrogen gas in the system during the entire process of the reaction was 1 standard atmospheric pressure, and the reaction mixture was reacted at 30 °C, the hydrogen atmosphere was 12 h. After the reaction is completed, the solvent is removed by rotating, then the pure target compound is obtained by column chromatography (eluent: petroleum ether / ethyl acetate = 6:1), yield: 93%. |
| 92% |
With sodium tetrahydridoborate In methanol at 25℃; for 16h; Cooling with ice; |
1.1 Step 1: Synthesis of 1-(pyridin-2-yl)ethanol.
To an ice-cooled solution of 1-(pyridin-2-yl)ethanone (11 g, 91 mmol) in methanol(120 mL) was slowly added sodium borohydride (8.58 g, 227 mmol). Upon completeaddition, the mixture was warmed to 25 °C. After 16 h, the reaction was diluted with water(100 mL), and the mixture was concentrated in vacuo. The resulting aqueous solution wasextracted with ethyl acetate (3 x 100 mL). The combined organic extracted were washed withsaturated aqueous sodium chloride (2 x 50 mL), dried over anhydrous sodium sulfate,filtered, and concentrated. Purification by flash column chromatography (1 0-30% ethyl acetate in petroleum ether) afforded 1-(pyridin-2-yl)ethanol (10.3 g, 92% yield). 1H NMR (400 MHz, Chloroform-d) ö: 8.54 (d, I = 4.8 Hz, 1 H), 7.72 - 7.68 (m, 1 H), 7.30 (d, I = 8.0 Hz, 1 H), 7.22-7.20 (m, 1 H), 4.93 -4.88 (m, 1 H), 4.39 (br s, 1 H), 1.52 (d, I = 6.4 Hz, 3 H). |
| 92% |
With (4-NHCpr)Triaz(NHP<SUP>i</SUP>Pr<SUB>2</SUB>)<SUB>2</SUB>Mn(CO)<SUB>2</SUB>Br; potassium-t-butoxide; hydrogen In toluene at 80℃; for 4h; Inert atmosphere; Autoclave; |
|
| 92.9% |
With C40H37ClN2PRuS(1+)*C24H20B(1-); isopropanol; potassium hydroxide at 82℃; for 2h; |
2.6. Typical procedure for transfer hydrogenation of ketones
General procedure: The mixture of a ketone (0.2 mmol) and base (0.08 mmol)containingthe catalyst (0.1 mol%) in 2-propanol (6 ml) was stirred at82 °C. After the reactionwas complete, diethyl ether could be addedto the mixture and extract the ruthenium complexes followed byfiltration and neutralized with 1N HCl, washed with water anddried over anhydrous Na2SO4. Conversion obtained is related to theresidual unreacted ketone. Percentage of conversionwas calculatedby using GC method of the crude mixture and compared with theauthentic samples. Acetone was identified as only by-product in allthe cases. As the catalyst is stable in all organic solvents and it canbe recovered and the work up process is also very simple for thiscatalytic system. |
| 92% |
Stage #1: 2-acetylpyridine With pyridine N-oxide; Triethoxysilane; C27H47FeP3Si In tetrahydrofuran at 50℃; for 6h; Schlenk technique;
Stage #2: With sodium hydroxide In tetrahydrofuran; methanol at 60℃; for 24h; Schlenk technique; |
|
| 91% |
Stage #1: 2-acetylpyridine With n-butyllithium; 1-(2-hydroxyethyl)-3-methyl-1H-imidazol-3-ium trifluoromethanesulfonate; ferrous acetate In tetrahydrofuran at 65℃; for 1h; Inert atmosphere;
Stage #2: With water monomer; sodium hydroxide In tetrahydrofuran; methanol at 20℃; for 2h; Inert atmosphere; |
|
| 91% |
With sodium tetrahydridoborate In methanol at 20℃; for 12h; |
|
| 91% |
With sodium tetrahydridoborate In methanol at 0 - 20℃; for 12h; |
|
| 90% |
With sodium tetrahydridoborate In ethanol for 2h; Heating; |
|
| 90% |
Stage #1: 2-acetylpyridine With C39H46ClCuN2; phenylsilane; potassium-t-butoxide In tetrahydrofuran at 20℃; for 0.416667h; Schlenk technique; Inert atmosphere; Glovebox;
Stage #2: With water monomer; sodium hydroxide In tetrahydrofuran; methanol for 1h; Schlenk technique; Glovebox; chemoselective reaction; |
|
| 90% |
With BH3; (R)-tetrahydro-1-methyl-3,3-diphenyl-1H,3H-pyrrolo[1,2-c]-[1,3,2]oxazaborole In tetrahydrofuran; 2-methyltetrahydrofuran at 0℃; Flow reactor; Green chemistry; |
|
| 89% |
With potassium hydroxide In tetrahydrofuran at 20℃; for 48h; Inert atmosphere; Schlenk technique; Glovebox; chemoselective reaction; |
|
| 89% |
Stage #1: 2-acetylpyridine With bis-[N,N′-bis(2,6-(di-isopropyl)phenyl)imidazol-2-ylidene]-(1H-1,2,4-triazol-1-yl)}copper(I) In tetrahydrofuran at 55℃; for 16h;
Stage #2: With sodium hydroxide In methanol; water monomer at 25℃; for 1.5h; |
|
| 89% |
With Mn(CO)<SUB>3</SUB>Br(k<SUP>2</SUP>P,N-Ph<SUB>2</SUB>PN(H)Py); hydrogen; 1,1,1,3,3,3-hexamethyldisilazane potassium In toluene at 80℃; for 20h; Glovebox; Autoclave; Inert atmosphere; |
|
| 89% |
Stage #1: 2-acetylpyridine With C36H36FeN6 at 20℃; for 22h; Inert atmosphere; Glovebox;
Stage #2: With water monomer; sodium hydroxide for 1.5h; |
|
| 87% |
With hydrogen In toluene at 25℃; for 8h; |
|
| 86% |
With methanol; sodium tetrahydridoborate at 20℃; for 4h; |
|
| 85% |
With dodecane; Triethoxysilane; hydridoiron(II) (trimethylphosphane)3(benzophenone imine) In tetrahydrofuran at 55℃; for 2h; Schlenk technique; Inert atmosphere; |
|
| 85% |
Stage #1: 2-acetylpyridine With bis(η5-cyclopentadienyl) titanium dichloride; sodium tetrahydridoborate In 1,2-dimethoxyethane at 20℃;
Stage #2: With sodium hydroxide In 1,2-dimethoxyethane |
|
| 85% |
With methanol; sodium tetrahydridoborate In tetrahydrofuran at 0℃; Inert atmosphere; |
|
| 83% |
Stage #1: 2-acetylpyridine With diphenylsilane; Cs2CO3 at 80℃; for 5h; Schlenk technique;
Stage #2: With sodium hydroxide In methanol; water monomer at 70℃; Schlenk technique; chemoselective reaction; |
|
| 82% |
With sodium tetrahydridoborate In ethanol at 0 - 20℃; |
|
| 80% |
With Triethoxysilane; C43H48FeP4 In tetrahydrofuran at 50℃; for 16h; |
|
| 80% |
With ammonia hydrochloride; zinc powder In tetrahydrofuran; water monomer at 60℃; for 4h; |
|
| 80% |
With C12H12MnO4(1+)*BF4(1-); potassium-t-butoxide In isopropanol at 90℃; for 24h; Schlenk technique; Inert atmosphere; |
|
| 80% |
With methanol; bis[dichlorido(η5-1,2,3,4,5-pentamethyl-cyclopentadienyl)iridium(III)]; 6,6′-dihydroxy-2,2′-bipyridine; potassium hydroxide at 60℃; for 24h; Inert atmosphere; |
|
| 79% |
With C56H55ClN3P2Ru(1+)*F6P(1-); potassium 2-methyl-2-butoxide In isopropanol at 20 - 80℃; for 1.5h; Schlenk technique; Inert atmosphere; |
|
| 79% |
With hydrogen; anhydrous silver perchlorate; 1,1,1,3,3,3-hexamethyldisilazane potassium In toluene at 25℃; for 17h; Glovebox; |
|
| 78% |
With potassium hydroxide; 18-crown-6 ether; hydrogen; isopropanol at 75℃; for 6h; |
|
| 78% |
Stage #1: 2-acetylpyridine With ferrous acetate; tricyclohexylphosphine In tetrahydrofuran at 65℃; Inert atmosphere;
Stage #2: In tetrahydrofuran at 65℃; Inert atmosphere;
Stage #3: With water monomer; sodium hydroxide In tetrahydrofuran; methanol at 0 - 20℃; Inert atmosphere; |
|
| 73% |
With cis-[(H)(SePh)Fe(PMe3)4]; sodium tertiary butoxide In isopropanol at 80℃; for 24h; |
|
| 72% |
Stage #1: 2-acetylpyridine With C40H32Cl2N3PRu; isopropanol at 82℃; for 0.166667h; Schlenk technique; Inert atmosphere;
Stage #2: With sodium hydroxide for 4h; Schlenk technique; Inert atmosphere; |
|
| 70% |
With sodium tetrahydridoborate In methanol at 20℃; for 5h; |
|
| 70% |
With sodium tetrahydridoborate In water monomer at 90℃; for 1.25h; |
Synthesis of α-methyl-2-pyridinemethanol.
General procedure: 20 g (0.17 mol) of 2-acetyl-pyridine were dissolved in 250 mL of EtOH. 12.5 g (0.3 mol) of NaBH4 were added slowly. After 1h at room temperature, the conversion of the substrate was complete. 250 mL of water were added and the solution was heated at 90°C for 15 min. After cooling, the product was extracted with AcOEt. The combined organic solutions were dried over MgSO4,the solvent was evaporated to give 13g (70%) of α-methyl-2-pyridine methanol as a pale green oil. 1H NMR (250 MHz, CDCl3): (ppm) = 1.49 (3H, d, J = 6 Hz); 4.94 (1H, q, J = 6 Hz); 7.21 (1H, m);7.28 (1H, m); 7.73 (1H, m); 8.48 (1H, m). Purity = 99% (NMR). |
| 70% |
Stage #1: 2-acetylpyridine With Triethoxysilane; [cis-Fe(H)(SPh)(PMe3)4] In tetrahydrofuran at 60℃; for 24h;
Stage #2: With methanol; sodium hydroxide In tetrahydrofuran; water monomer at 60℃; for 24h; |
2.2. General procedure for catalytic hydrosilylation of aldehydes
General procedure: To a 25 mL Schlenk tube containing a solution of 1 in 2 mL of THF was added an aldehyde (1.0 mmol) and (EtO)3 SiH (0.20 g, 1.2 mmol). The reaction mixture was stirred at 50-55 °C until there was no aldehyde left (monitored by TLC and GC-MS). The reaction was then quenched byMeOH (2mL) and a 10% aqueous solution of NaOH (5 mL) with vigorous stirring at 60 °C for about 24 h.The organic product was extracted with diethyl ether (10 mL × 3), dried over anhydrous MgSO4, and concentrated under vacuum. The alcohol product was further purified using flash column chromatography (elute with 5-10% ethyl acetate in petroleum ether). The 1H NMR and 13C NMR spectra of the alcohol products are providedin Supporting information. |
| 69% |
With sodium tetrahydridoborate In methanol; dichloromethane at 20℃; for 6h; |
|
| 66% |
With Triethoxysilane; C27H42FeP4S In tetrahydrofuran at 50℃; for 24h; Schlenk technique; Green chemistry; |
|
| 63% |
With dicarbonyl-(2,4-bis(trimethylsilyl)bicyclo[3.3.0]nona-1,4-dien-3-one)[acetonitrile]iron; isopropanol at 80℃; for 18h; Inert atmosphere; |
|
| 56.1% |
With methanol; sodium tetrahydridoborate at 20℃; for 18h; |
35 1-Pyridin-2-yl-ethanol
1-Pyridin-2-yl-ethanol 2-Acetylpyridine (1.0 g, 8.26 mmol) was dissolved in anhydrous methanol (20 mL) and treated with sodium borohydride (0.62 g, 16.51 mmol) and stirred at ambient temperature 18 hours. A saturated solution of ammonium chloride was added and the mixture was extracted with ethyl acetate. The combined organics were washed with water and brine, dried over anhydrous sodium sulfate, evaporated and chromatographed using a gradient of 50 to 100% ethyl acetate in hexanes to provide 0.57 g (56.1%) of the title compound. MS m/z 124 (M+H)+. |
| 50% |
With anhydrous magnesium perchlorate; diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate In acetonitrile at 70℃; for 168h; |
|
| 37% |
With C51H46N3OP2Ru; potassium hydroxide In isopropanol at 80℃; for 2h; |
2.5. Catalysis: general procedure for the transfer hydrogenationreactions
General procedure: A mixture of ketone (1 mmol), a known mole percent of the catalystand KOH (0.06 mmol) was dissolved in 2-propanol (5 mL), and the mixture was heated under reflux for the desired period oftime. The catalyst was removed as precipitate from the reactionmixture by the addition of diethyl ether followed by filtrationand subsequent neutralization with 5 mL of 1 M HCl. Then theether layer was passed through a short path of silica gel and the filtratewas subjected to GC analysis for identification andquantification. |
| 35% |
Stage #1: 2-acetylpyridine With (dppe)2Fe(H)2*(C7H8)2; Na-tetrakis(ethoxy)borate In toluene at 100℃; for 2h; visible light irradiation; Inert atmosphere;
Stage #2: With water monomer; sodium hydroxide In methanol; toluene at 20℃; for 16h; |
|
| 20% |
With sodium tetrahydridoborate In methanol at 20℃; |
|
|
With hydrogenchloride; zinc,6a mercury,6b |
|
|
With sodium tetrahydridoborate Yield given; |
|
|
With sodium tetrahydridoborate In methanol Ambient temperature; |
|
|
With sodium tetrahydridoborate |
|
|
With sodium tetrahydridoborate In methanol for 0.5h; |
|
|
With sodium tetrahydridoborate |
|
|
Stage #1: 2-acetylpyridine With sodium tetrahydridoborate In methanol at 0℃; for 4h;
Stage #2: With methanol; water monomer |
11.1
Production Example 11-1: 1-(Pyridin-2-yl)ethanol: A methanol solution (100 mL) of 1-pyridin-2-ylethanone (10.0 g) was cooled to 0°C, sodium borohydride (6.2 g) was added thereto and stirred at that temperature for 4 hours. Then water was added to the reaction solution, and the organic solvent was evaporated off under reduced pressure. The resulting residue was extracted with ethyl acetate. The organic layer was dried with anhydrous magnesium sulfate, and the solvent was evaporated off under reduced pressure. The resulting residue was purified through silica gel column chromatography (hexane/ethyl acetate = 4/1 to 3/2) to obtain the entitled compound (9.0 g). |
|
With potassium hydroxide; Au/TiO2; isopropanol at 82℃; for 8h; Inert atmosphere; chemoselective reaction; |
|
|
With ethanol; CF3O3S(1-)*C48H38NO3PRh(1+); potassium carbonate at 40℃; |
|
|
With sodium tetrahydridoborate In ethanol Inert atmosphere; |
|
| 99 %Spectr. |
With hydrogen In 1-butyl-3-methylimidazolium hexafluorophosphate at 20℃; for 24h; |
General procedure: The palladium nanoparticles on SWNTs were prepared in [BMIM][PF6]. The SWNTs (5 mg) was grounded in IL (1 ml) for 30 min, and then Pd(II) acetate (0.018 mmol) was dissolved in the solution. The Pd(II) acetate was in situ reduced in IL with 1 atm of hydrogen for 5 min at room temperature. The aryl ketone (0.3 mmol) was added to this solution under 1 atm of hydrogen at room temperature. After the >99% completion of the reaction was checked by TLC, the products were extracted with ethyl ether. The ethereal phase was concentrated and analyzed by 1H NMR. |
| 98 %Spectr. |
With potassium fluoride; palladium diacetate; chlorobenzene In tetrahydrofuran; water monomer at 20℃; for 1h; Inert atmosphere; chemoselective reaction; |
|
| 87 %Chromat. |
With [(iPrPNP)FeH(CO)Br]; potassium-t-butoxide; hydrogen In ethanol at 26 - 28℃; for 15h; |
|
|
With Ru(P,O,P-xantphos)(S-dmso)Cl2; isopropanol; potassium hydroxide for 12h; Reflux; |
|
| 18 %Chromat. |
With anhydrous sodium carbonate; isopropanol at 82℃; for 8h; Inert atmosphere; |
|
| 99 %Chromat. |
With C71H56N5P2Ru(1+)*Cl(1-); potassium isopropoxide; isopropanol at 82℃; for 6h; Inert atmosphere; |
|
| 99 %Chromat. |
Stage #1: 2-acetylpyridine With [RuCl2(η6-benzene)tris(4-methoxyphenyl)phosphane] In isopropanol at 82℃; for 0.166667h; Inert atmosphere;
Stage #2: With potassium isopropoxide In isopropanol at 82℃; for 9h; Inert atmosphere; |
|
|
Multi-step reaction with 2 steps
1: C24H50FeO2P4 / tetrahydrofuran / 36 h / 50 °C / Inert atmosphere
2: water monomer; sodium hydroxide / tetrahydrofuran; methanol / 48 h / 50 °C |
|
| > 99 %Chromat. |
With acetonitrile(η5-pentamethylcyclopentadienyl)(κ2-C,N-3-methyl-1-(2-picolyl)imidazol-2-ylidene)ruthenium(II) hexafluorophosphate; isopropanol; potassium hydroxide at 82℃; for 3h; Inert atmosphere; |
|
|
With sodium tetrahydridoborate In methanol at 60℃; for 6h; |
|
|
With C55H41ClN2O3P2Ru; isopropanol; potassium hydroxide In 1,3-dimethylbenzene at 20 - 82℃; for 4h; Inert atmosphere; |
Typical procedure for transfer hydrogenation of ketones
General procedure: In an oven-dried round bottom flask, were placed ketone (2.4 mmol), catalyst (3 mol), base (12 mol), internal standard (m-xylene, 30 L, 0.24 mmol) and i-PrOH (5 mL) at room temperature. The reaction mixture was heated at 82 °C for the required reaction time under an atmosphere of nitrogen. Aliquots (0.2 mL) were taken at fixed time and the catalyst removed as precipitate from the reaction mixture by the addition of diethyl ether. The organic layer was neutralized with 1 N HCl, washed with water and dried over anhydrous Na2SO4. The combined organic layer passed through a short path of silica gel and then subjected to GCMS analysis. The conversions obtained are related to the residual unreacted ketone and are averages of two runs in the case of all catalytic reactions. |
|
With sodium tetrahydridoborate |
|
| 79 %Spectr. |
With trans-RuCl(6,6'-dihydroxyterpyridine)(PPh<SUB>3</SUB>)<SUB>2</SUB>PF<SUB>6</SUB>; potassium-t-butoxide; isopropanol at 80℃; for 24h; Inert atmosphere; Sealed tube; chemoselective reaction; |
|
| 99 %Spectr. |
With C24H20B(1-)*C40H43NO2PRuS(1+); isopropanol; potassium hydroxide at 80℃; for 12h; Inert atmosphere; |
4.5 Typical procedure for the catalytic transfer hydrogenation of ketones
General procedure: In a dry two-necked round bottom flask under an atmosphere of nitrogen were placed an appropriate amount of catalyst 1-5 (0.01 mmol), (0.025 mmol) of KOH and (10 mmol) of aryl ketone in 2-propanol (10 ml) was added and the resulting mixture was refluxed under an atmosphere of nitrogen and the course of the reaction was monitored by 1H NMR analysis. After completion of the reaction, the solvent was removed under reduced pressure. The catalyst was removed by the addition of 15 ml of ether (b.p., 40-60 °C) followed by filtration and subsequent neutralization with dilute HCl. The combined organic fractions were dried over anhydrous Na2SO4. The solvent was distilled off to obtain a crude mixture containing ketone and its hydrogenated product. Percentage conversion was calculated by 1H NMR spectra of the crude mixture. The only side product formed is acetone, which is easily removed by distillation during workup. |
|
With [(bis(5-methyl-3-phenyl-1,2,4-triazolyl)(3-methyl-5-phenyl-1,2,4-triazolyl)borate)Ru(p-cymene)Cl]; potassium hydroxide In isopropanol for 24h; Inert atmosphere; Reflux; |
|
|
Multi-step reaction with 2 steps
1: mer-hydrido(2-mercaptobenzoyl)tris(trimethylphosphine)cobalt(III) / tetrahydrofuran / 8 h / 55 °C / Inert atmosphere
2: sodium hydroxide / tetrahydrofuran; water monomer / 56 h / 50 °C / Inert atmosphere |
|
|
With methanol In toluene at 20℃; for 4.5h; Inert atmosphere; Sealed tube; Irradiation; |
|
|
With RuCl(CH3(C6H4)CH3CHCH3)((C5H4N)C2N3(CH3)2)(1+)*CF3SO3(1-) = [RuCl(CH3(C6H4)C3H7)((C5H4N)C2N3(CH3)2)](CF3SO3); potassium hydroxide In isopropanol for 24h; Reflux; |
|
|
With potassium-t-butoxide In isopropanol at 90℃; for 0.533333h; Flow reactor; Inert atmosphere; |
|
| 99 %Spectr. |
With [Fe(N,N′-bis(diisopropylphosphino)-2,6-diaminopyridine)(H)(CO)Br]; potassium-t-butoxide; hydrogen In ethanol at 25℃; for 1h; |
|
|
With C54H41Cl2N2O2P2Ru; isopropanol; potassium hydroxide at 82℃; for 5h; |
Typical procedure for transfer hydrogenation of ketones
General procedure: The mixture of a ketone (0.2 mmol) and base (0.08 mmol) containing the catalyst (0.002 mmol) in i-PrOH (6 ml) was stirred at 82 °C. After the reaction was complete, diethyl ether could be added to the mixture and extract the ruthenium complexes followed by filtration and neutralized with 1 N HCl, washed with water and dried over anhydrous Na2SO4. Conversion obtained is related to the residual unreacted ketone. The alcohol products were identified by comparison with the authentic samples. Acetone was identified as only by-product in all the cases. As the catalyst is stable in all organic solvents and it can be recovered and the work up process is also very simple for this catalytic system. |
|
With 1-butyl-2,3-dimethylimidazolium hydroxide; hydrogen at 30℃; for 15h; Autoclave; chemoselective reaction; |
|
|
With C10H14Cl3Ru(1-)*C19H15Cl3N3(1+)*H2O; potassium hydroxide In isopropanol at 82℃; for 4h; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping