
Lactones with Methylcyclohexane Systems Obtained by Chemical and Microbiological Methods and Their Antimicrobial Activity

 and
and
Abstract
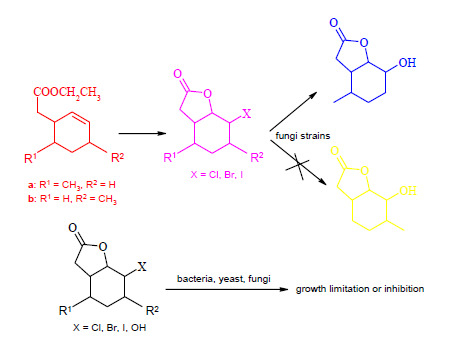
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Substrates

2.2. Biotransformation of Halolactones

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
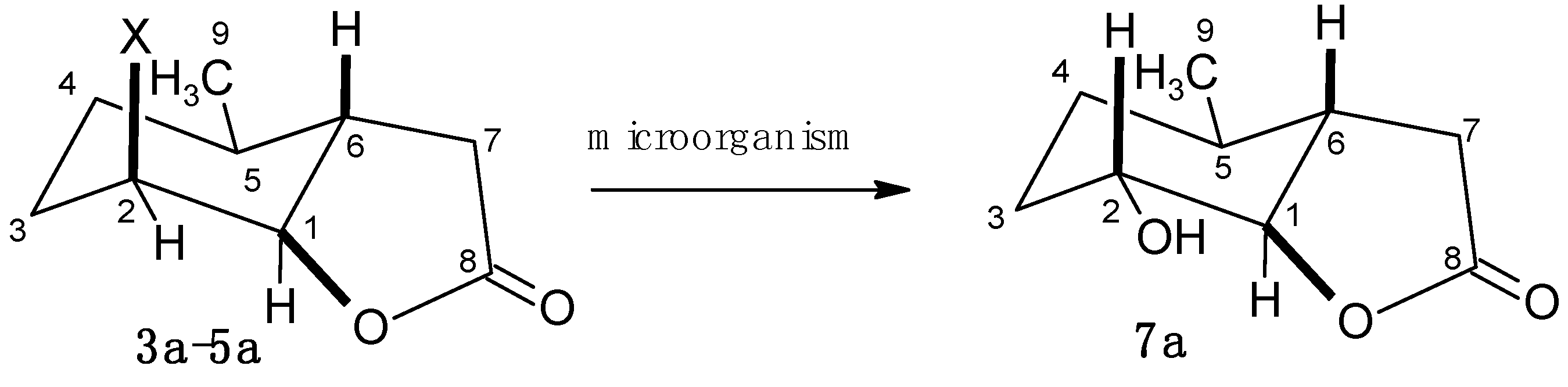{kind=link}
| Entry | Microorganism | Chlorolactone | Bromolactone | Iodolactone | |||
|---|---|---|---|---|---|---|---|
| 3a | 3b | 4a | 4b | 5a | 5b | ||
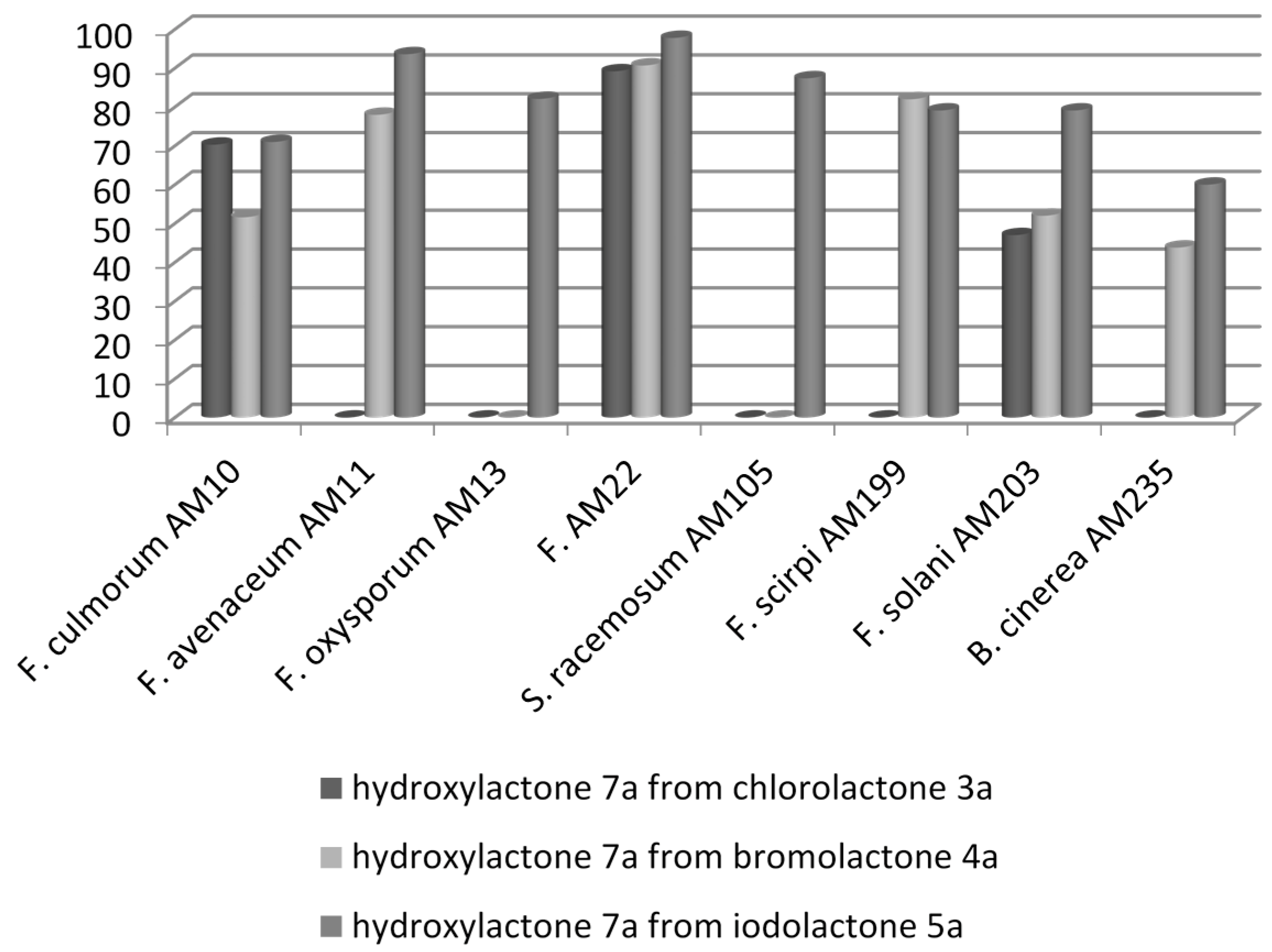
| 1 | Fusarium culmorum AM10 | (++) | (‒) | (++) | (‒) | (+++) | (‒) |
| 2 | Fusarium avenaceum AM11 | (‒) | (‒) | (+++) | (‒) | (++++) | (‒) |
| 3 | Fusarium oxysporum AM13 | (‒) | (‒) | (+) | (‒) | (+++) | (‒) |
| 4 | Fusarium tricinctum AM16 | (‒) | (‒) | (‒) | (‒) | (‒) | (‒) |
| 5 | Fusarium semitectum AM20 | (‒) | (‒) | (+) | (‒) | (+) | (‒) |
| 6 | Fusarium equiseti AM22 | (+++) | (‒) | (++++) | (‒) | (++++) | (‒) |
| 7 | Syncephalastrum racemosum AM105 | (‒) | (‒) | (‒) | (‒) | (+++) | (‒) |
| 8 | Fusarium scirpi AM199 | (‒) | (‒) | (+++) | (‒) | (+++) | (‒) |
| 9 | Fusarium solani AM203 | (++) | (‒) | (++) | (‒) | (+++) | (‒) |
| 10 | Botrytis cinerea AM235 | (‒) | (‒) | (‒) | (‒) | (++) | (‒) |


| Entry | Strain | Yield (g)/(%) | ee | |
|---|---|---|---|---|
| 1 | F. culmorum AM10 | 0.017/18.3 | 45.4 | ‒33.344 (c = 0.69, CHCl3) |
| 2 | F. equiseti AM22 | 0.038/41.8 | 34.3 | ‒23.175 (c = 0.88, CHCl3) |
| 3 | F. solani AM203 | 0.012/13.3 | 17.9 | +14.509 (c = 0.46, CHCl3) |
| Entry | Strain | Yield (g)/(%) | ee | |
|---|---|---|---|---|
| 1 | F. culmorum AM10 | 0.024/32.4 | 32.4 | ‒22.477 (c = 0.85, CHCl3) |
| 2 | F. avenaceum AM11 | 0.032/10.4 | 10.4 | +9.765 (c = 1.65, CHCl3) |
| 3 | F. equiseti AM22 | 0.024/18.0 | 32.5 | ‒17.042 (c = 0.61, CHCl3) |
| 4 | F. scirpi AM199 | 0.028/41.2 | 38.5 | ‒27.339 (c = 0.87, CHCl3) |
| 5 | F. solani AM203 | 0.034/5.6 | 46.6 | +4.234 (c = 0.85, CHCl3) |
| 6 | B. cinerea AM235 | 0.021/20.2 | 28.9 | ‒19.213 (c = 0.94, CHCl3) |
| Entry | Strain | yield (g)/(%) | ee | |
|---|---|---|---|---|
| 1 | F. culmorum AM10 | 0.016/26.7 | 45.2 | ‒32.244 (c = 0.89, CHCl3) |
| 2 | F. avenaceum AM11 | 0.018/29.7 | 5.7 | ‒6.598 (c = 0.80, CHCl3) |
| 3 | F. oxysporum AM13 | 0.035/57.2 | 35.3 | +37.211 (c = 0.59, CHCl3) |
| 4 | F. equiseti AM22 | 0.036/58.5 | 15.1 | ‒17.740 (c = 0.97, CHCl3) |
| 5 | S. racemosum AM105 | 0.031/50.7 | 24.0 | ‒19.714 (c = 0.48, CHCl3) |
| 6 | F. scirpi AM199 | 0.030/49.6 | 47.6 | ‒34.134 (c = 0.65, CHCl3) |
| 7 | F. solani AM203 | 0.013/21.4 | 3.7 | 0 (c = 0.72, CHCl3) |
| 8 | B. cinerea AM235 | 0.014/23.2 | 45.2 | ‒32.196 (c = 0.49, CHCl3) |
2.3. Biological Tests
| Entry | Strain | OD Control | max.OD of 3a | max.OD of 4a | max.OD of 5a | max.OD of 6a | max.OD of 7a |
|---|---|---|---|---|---|---|---|
| 1 | E. coli | 1.56 | 1.53 a | 1.57 a | 1.46 a | 1.50 a | 1.50 |
| 2 | S. aureus | 1.55 | 1.20 | 1.56 | 1.58 | 1.22 | 1.45 |
| 3 | B. subtilis | 1.37 | 1.39 b | 1.38 b | 1.40 b | 1.40 | 1.56 |
| 4 | C. albicans | 1.96 | 1.56 c | 0.41 | 0.95 | 1.80 c | 1.90 |
| 5 | S. cerevisiae | 2.30 | 0.80 | 0.70 | 1.37 | 1.67 | 0.37 |
| 6 | Y. lipolytica | 1.40 | 0.38 | 0.49 | 0.89 | 1.05 c | 1.41 |
| 7 | F. linii | 1.96 | 0.32 | 0.36 | 0.45 | 1.51 | 2.45 |
| 8 | A. niger | 3.0 | 2.00 d | 0.81 | 1.22 | 1.16 | 2.71 |
| Entry | Strain | OD Control | Max.OD of 3b | Max.OD of 4b | Max.OD of 5b |
|---|---|---|---|---|---|
| 1 | E. coli | 1.56 | 1.56 | 0.72 | 1.46 |
| 2 | S. aureus | 1.55 | 1.00 | 0.84 | 1.30 |
| 3 | B. subtilis | 1.4 | 1.00 a | 1.20 a | 1.20 |
| 4 | C. albicans | 1.96 | 1.82 | 1.85 | 0.58 |
| 5 | S. cerevisiae | 2.30 | 0.70 | 0.70 | 1.11 |
| 6 | Y. lipolytica | 1.39 | 0.39 | 0.36 | 1.00 b |
| 7 | F. linii | 2.05 | 0.35 | 0.37 | 0.31 |
| 8 | A. niger | 3.0 | 0.48 | 0.36 | 2.41 c |
2.4. Discussion
2.4.1. Characterization of Compounds 3a–6a with 5-Methylcyclohexane Rings



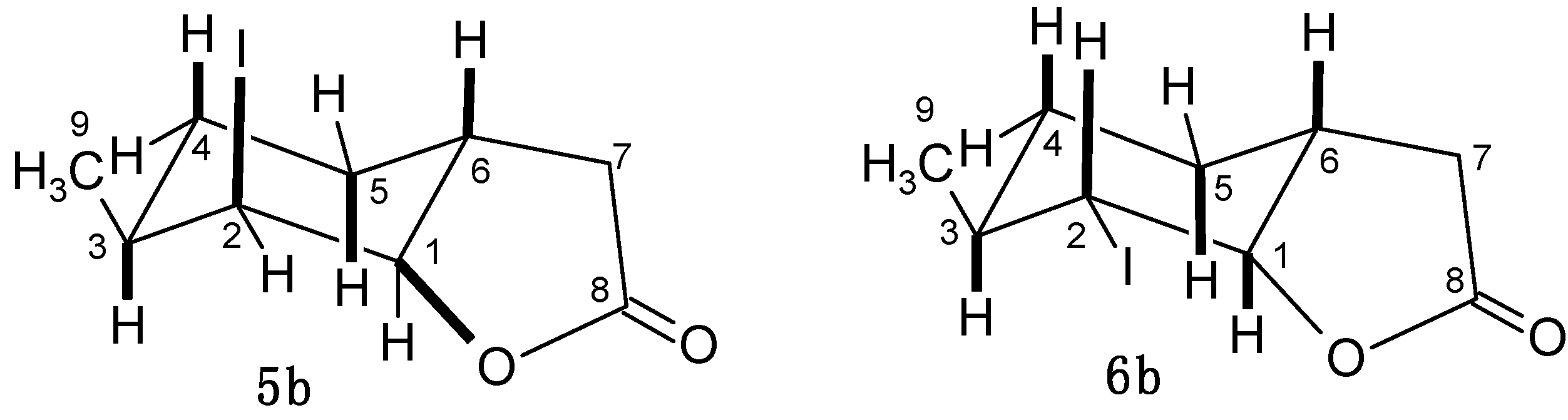
2.4.2. Characterization of Compounds with 3-Methylcyclohexane Ring (3b–6b)

2.4.3. The Assessment of the Effects of Lactones on the Growth of Tested Microorganisms


2.4.4. An olfactory Analysis of Lactones
- acid 2a—medium intensive, sweet-sour, dry pine scent;
- chlorolactone 3b—slight dried fruit scent;
- iodolactone 5b—slight scent of dust, reminiscent of an old pharmacy.
3. Experimental Section
3.1. General
3.2. Synthesis of Substrates
3.2.1. (6–Methylcyclohex-2-en-1-yl)acetic Acid (2a)
3.2.2. 2-Chloro-5-methyl-9-oxabicyclo[4.3.0]nonan-8-one (3a)
3.2.3. 2-Bromo-5-methyl-9-oxabicyclo[4.3.0]nonan-8-one (4a)
3.2.4. 2-Iodo-5-methyl-9-oxabicyclo[4.3.0]nonan-8-one (5a) and 2-Iodo-5-methyl-9-oxabicyclo[4.3.0]-nonan-8-one (6a)
3.2.5. (4–Methylcyclohex-2-en-1-yl)acetic Acid (2b)
3.2.6. 2-Chloro-3-methyl-9-oxabicyclo[4.3.0]nonan-8-one (3b)
3.2.7. 2-Bromo-3-methyl-9-oxabicyclo[4.3.0]nonan-8-one (4b)
3.2.8. 2-Iodo-3-methyl-9-oxabicyclo[4.3.0]nonan-8-one (5b) and 2-Iodo-3-methyl-9-oxabicyclo-[4.3.0]nonan-8-one (6b)
3.3. Biotransformations
3.3.1. Microorganisms
3.3.2. Bioassays
3.3.3. Odour Evaluation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, H.; Xu, C.; Liu, D.Q.; An, S.Q.; Tan, R.X. Buddlin, a new compound from Buddleja asiatica. Fitoterapia 2005, 76, 588–589. [Google Scholar] [CrossRef] [PubMed]
- Pujar, P.P.; Sawaikar, D.D.; Rojatkar, S.R.; Nagasampagi, B.A. A new germacranolide from Artemisia pallens. Fitoterapia 2000, 71, 590–592. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Khattab, A.M.; Grace, M.H.; Sahl, M.M. A new eudesmanolide from Crataegus flava fruits. Fitoterapia 2001, 72, 756–759. [Google Scholar] [CrossRef]
- Ortet, R.; Prado, S.; Mouray, E.; Thomas, O.P. Sesquiterpene lactones from the endemic Cape Verdean Artemisia gorgonom. Phytochemistry 2008, 69, 2961–2965. [Google Scholar] [CrossRef] [PubMed]
- Marco, J.A.; Sanz-Cervera, J.F.; Yuste, A.; Sancenon, F.; Carda, M. Sesquiterpenes from Centaurea aspera. Phytochemistry 2005, 66, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Xu, Y.J.; Liu, H.L.; Liu, X.Q.; Shang, J.N.; Han, G.T.; Yao, M.J.; Yuan, C.S. Eremophilane-type sesquiterpene lactones from Ligularia hodgsonii Hook. Phytochemistry 2011, 72, 514–517. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.S.; Zheng, Y.L.; Mo, J.X.; Liu, X.; Li, X.H.; Zhou, C.X. Sesquiterpene lactones from the root tubers of Lindera agregata. J. Nat. Prod. 2009, 72, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Buskuhl, H.; de Oliveira, F.L.; Blind, L.Z.; de Freitas, R.A.; Barison, A.; Campos, F.R.; Corilo, Y.E.; Eberlin, M.N.; Caramori, G.F.; Biavatti, M.W. Sesquiterpene lactones from Vernonia scorpioides and their in vitro cytotoxicity. Phytochemistry 2010, 71, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, A.M.; Juarez, Z.N.; Bach, H.; Nematallah, A.; Av-Gay, Y.; Sanchez-Arreola, E.; Catalan, C.A.N.; Turbay, S.; Hernandez, L.R. Antimicrobial activities of sesquiterpene lactones and inositol derivatives from Hymenoxys robusta. Phytochemistry 2011, 72, 2413–2418. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Lantvit, D.D.; Riswan, S.; Kardono, L.B.S.; Chai, H.B.; de Blanco, E.J.C.; Farnsworth, N.R.; Soejarto, D.D.; Swanson, S.; Kinghorn, A.D. Bioactivity-guided isolation of cytotoxic sesquiterpenes of Rolandra fruticosa. Phytochemistry 2010, 71, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Zhang, M.; Zhang, C.F.; Qi, H.Y.; Bashall, A.; Bligh, S.W.A.; Wang, Z.T. Cytotoxic sesquiterpenes from Ligularia platyglossa. Phytochemistry 2008, 69, 2231–2236. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Noh, H.J.; Choi, S.U.; Park, K.M.; Seok, S.J.; Lee, K.R. Lactarane sesquiterpenoids from Lactarius subvellereus and their cytotoxicity. Bioorg. Med. Chem. Lett. 2010, 20, 5385–5388. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ahn, J.H.; Kim, S.B.; Lee, C.; Hwang, B.Y.; Lee, M.K. Sesquiterpene lactones from the roots of Lindera strychnifolia. Phytochemistry 2013, 87, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Komala, I.; Ito, T.; Nagashima, F.; Yagi, Y.; Kawahata, M.; Yamaguchi, K.; Asakawa, Y. Zierane sesquiterpene lactone, cembrane and fusicoccane diterpenoids, from the Tahitian liverwort Chandonanthus hirtellus. Phytochemistry 2010, 71, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Pillay, P.; Vleggaar, R.; Maharaj, V.J.; Smith, P.J.; Lategan, C.A. Isolation and identification of antiplasmodial sesquiterpene lactones from Oncosiphon piluliferum. J. Ethnopharmacol. 2007, 112, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.M.S.; Theodoro, P.N.E.T.; Eparvier, V.; Basset, C.; Silva, M.R.R.; Beauchene, J.; Espindola, L.S.; Stien, D. Search for antifungal compounds from the wood of durable tropical trees. J. Nat. Prod. 2010, 73, 1706–1707. [Google Scholar] [CrossRef] [PubMed]
- Yesilada, E.; Gurbuz, I.; Bedir, E.; Tatli, I.; Khan, I.A. Isolation of anti-ulcerogenic sesquiterpene lactones from Centaurea solstitialis L. ssp. solstitialis through bioassay-guided fractionation procedures in rats. J. Ethnopharmacol. 2004, 95, 213–219. [Google Scholar]
- Xiao, W.; Li, X.; Li, N.; Bolati, M.; Wang, X.; Jia, X.; Zhao, Y. Sesquiterpene lactones from Saussurea involucrate. Fitoterapia 2011, 82, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Evidente, A.; Masi, M.; Linaldeddu, B.T.; Franceschini, A.; Scanu, B.; Cimmino, A.; Andolfi, A.; Motta, A.; Maddau, L. Afritoxinones A and B, dihydrofuropyran-2-ones produced by Diplodia africana the causal agent of branch dieback on Juniperus phoenicea. Phytochemistry 2012, 77, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Sumioka, H.; Harinantenaina, L.; Matsunami, K.; Otsuka, H.; Kawahata, M.; Yamaguchi, K. Linderolides A–F, eudesmane-type sesquiterpene lactones and linderoline, a germacrane-type sesquiterpene from the roots of Lindera strychnifolia and their inhibitory activity on NO production in RAW 264.7 cells in vitro. Phytochemistry 2011, 72, 2165–2171. [Google Scholar] [CrossRef] [PubMed]
- Wube, A.A.; Wenzig, E.M.; Gibbons, S.; Asres, K.; Bauer, R.; Bucar, F. Constituents of the stem bark of Discopodium penninervium and their LTB4 and COX-1 and -2 inhibitory activities. Phytochemistry 2008, 69, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Koshimura, M.; Utsukihara, T.; Kawamoto, M.; Saito, M.; Horiuchi, C.A.; Kuniyoshi, M. Biotransformation of bromosesquiterpenes by marine fungi. Phytochemistry 2009, 70, 2023–2026. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.H.; Chung, H.M.; Hwang, T.L.; Lu, M.C.; Wen, Z.H.; Kuo, Y.H.; Wang, W.H.; Sung, P.J. Echinoclerodane A: A new bioactive clerodane-type diterpenoid from a gorgonian coral Echinomuricea sp. Molecules 2012, 17, 9443–9450. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Geng, C.A.; Ma, Y.B.; Zhang, X.M.; Chen, J.J. Three new secoiridoids, swermacrolactones A–C and anti-hepatitis B virus activity from Swertia macrosperma. Fitoterapia 2013, 89, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Guan, X.L.; Li, J.; Deng, S.P.; Li, L.Q.; Tang, M.T.; Huang, J.G.; Chen, Z.Z.; Yang, R.Y. Hypoglycemic effects and constituents of the barks of Cyclocarya paliurus, and their inhibiting activities to glucosidase and glycogen phosphorylase. Fitoterapia 2011, 82, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, X.W. Five new eudesmane-type sesquiterpenoid lactones biotransformed from atractylenolide I by rat hepatic microsomes. Fitoterapia 2013, 85, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Wedge, D.E.; Galindo, J.C.G.; Macias, F.A. Fungicidal activity of natural and synthetic sesquiterpene lactone analogs. Phytochemistry 2000, 53, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Aranda, G.; Moreno, L.; Cortes, M.; Prange, T.; Maurs, M.; Azerad, D. A new example of 1a-hydroxylation of drimanic terpenes through combined microbial and chemical processes. Tetrahedron 2001, 57, 6051–6056. [Google Scholar] [CrossRef]
- Maurs, M.; Azerad, R.; Cortes, M.; Aranda, G.; Delahaye, M.B.; Ricard, L. Microbial hydroxylation of natural drimenic lactones. Phytochemistry 1999, 52, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Gładkowski, W.; Mazur, M.; Białońska, A.; Wawrzeńczyk, C. Lactones 35. Metabolism of iodolactones with cyclohexane ring in Absidia cylindrospora culture. Enzyme Microb. Technol. 2011, 48, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Grabarczyk, M.; Białońska, A. Biotransformations of chloro-, bromo- and iodolactone with trimethylcyclohexane system using fungal strains. Biocatal. Biotransform. 2010, 28, 408–414. [Google Scholar] [CrossRef]
- Grotowska, A.K.; Wawrzeńczyk, C. Lactones 13. Biotransformation of iodolactones. J. Mol. Catal. B 2002, 19–20, 203–208. [Google Scholar] [CrossRef]
- Grabarczyk, M. Fungal strains as catalysts for the biotransformation of halolactones by hydrolytic dehalogenation with the dimethylcyclohexane system. Molecules 2012, 17, 9741–9753. [Google Scholar] [CrossRef] [PubMed]
- Grabarczyk, M.; Mączka, W.; Wińska, K.; Żarowska, B.; Anioł, M. Antimicrobial activity of hydroxylactone obtained by biotransformation of bromo- and iodolactone with gem-dimethylcyclohexane ring. J. Braz. Chem. Soc. 2013, 24, 1913–1919. [Google Scholar]
- Żołnierczyk, A.K.; Anioł, M.; Wawrzeńczyk, C. Microbial dehalogenation of chloro- and bromolactones. Przem. Chem. 2013, 92, 802–805. [Google Scholar]
- Grabarczyk, M.; Wińska, K.; Mączka, W.; Anioł, M. Synthesis and odour characteristics of hydroxylactones with methylcyclohexane system. Przem. Chem. 2014, 93, 1000–1003. [Google Scholar]
- Lebrun, M.E.; Pfeiffer, J.Y.; Beauchemin, A.M. Synthesis of 2-epi-pumiliotoxin C via a challenging intramolecular hydroamination key step. Synlett 2009, 7, 1087–1090. [Google Scholar]
- Semmelhack, M.F.; Epa, W.R.; Cheung, A.W.H.; Gu, Y.; Kim, C.; Zhang, N.; Lew, W. Palladium-promoted synthesis of ionophore antibiotics. Strategy and assembly of the homochiral tetrahydrofuran and tetrahydropyran portions of tetronomycin. J. Am. Chem. Soc. 1994, 116, 7455–7456. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Szumny, A.; Gładkowski, W.; Białońska, A.; Ciunik, Z.; Wawrzeńczyk, C. Lactones 18. Synthesis of bicyclic lactones with methyl-, di- and trimethyl substituted cyclohexane system. Pol. J. Chem. 2005, 79, 1763–1771. [Google Scholar]
- Snider, B.B.; Johnston, M.I. Regioselectivity of the halolactonization of γ,δ-unsaturated acids. Tetrahedron Lett. 1985, 26, 5497–5500. [Google Scholar] [CrossRef]
- Ranganathan, S.; Muraleedharan, K.M.; Vaish, N.K.; Jayaraman, N. Halo- and selenolactonisation: The two major strategies for cyclofunctionalisation. Tetrahedron 2004, 60, 5273–5308. [Google Scholar] [CrossRef]
- Janssen, D.B. Evolving haloalkane dehalogenases. Curr. Opin. Chem. Biol. 2004, 8, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Medina-Martínez, M.S.; Uyttendaele, M.; Rajkovic, A.; Nadal, P.; Debevere, J. Degradation of N-acyl-l-homoserine lactones by Bacillus cereus in culture media and pork extract. Appl. Environ. Microbiol. 2007, 73, 2329–2332. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 1a, b–6a, b are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabarczyk, M.; Wińska, K.; Mączka, W.; Żołnierczyk, A.K.; Żarowska, B.; Anioł, M. Lactones with Methylcyclohexane Systems Obtained by Chemical and Microbiological Methods and Their Antimicrobial Activity. Molecules 2015, 20, 3335-3353. https://doi.org/10.3390/molecules20023335
Grabarczyk M, Wińska K, Mączka W, Żołnierczyk AK, Żarowska B, Anioł M. Lactones with Methylcyclohexane Systems Obtained by Chemical and Microbiological Methods and Their Antimicrobial Activity. Molecules. 2015; 20(2):3335-3353. https://doi.org/10.3390/molecules20023335
Chicago/Turabian StyleGrabarczyk, Małgorzata, Katarzyna Wińska, Wanda Mączka, Anna K. Żołnierczyk, Barbara Żarowska, and Mirosław Anioł. 2015. "Lactones with Methylcyclohexane Systems Obtained by Chemical and Microbiological Methods and Their Antimicrobial Activity" Molecules 20, no. 2: 3335-3353. https://doi.org/10.3390/molecules20023335