
Lasianosides F–I: A New Iridoid and Three New Bis-Iridoid Glycosides from the Leaves of Lasianthus verticillatus (Lour.) Merr.

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Isolation and Spectroscopic Analyses of the Compounds
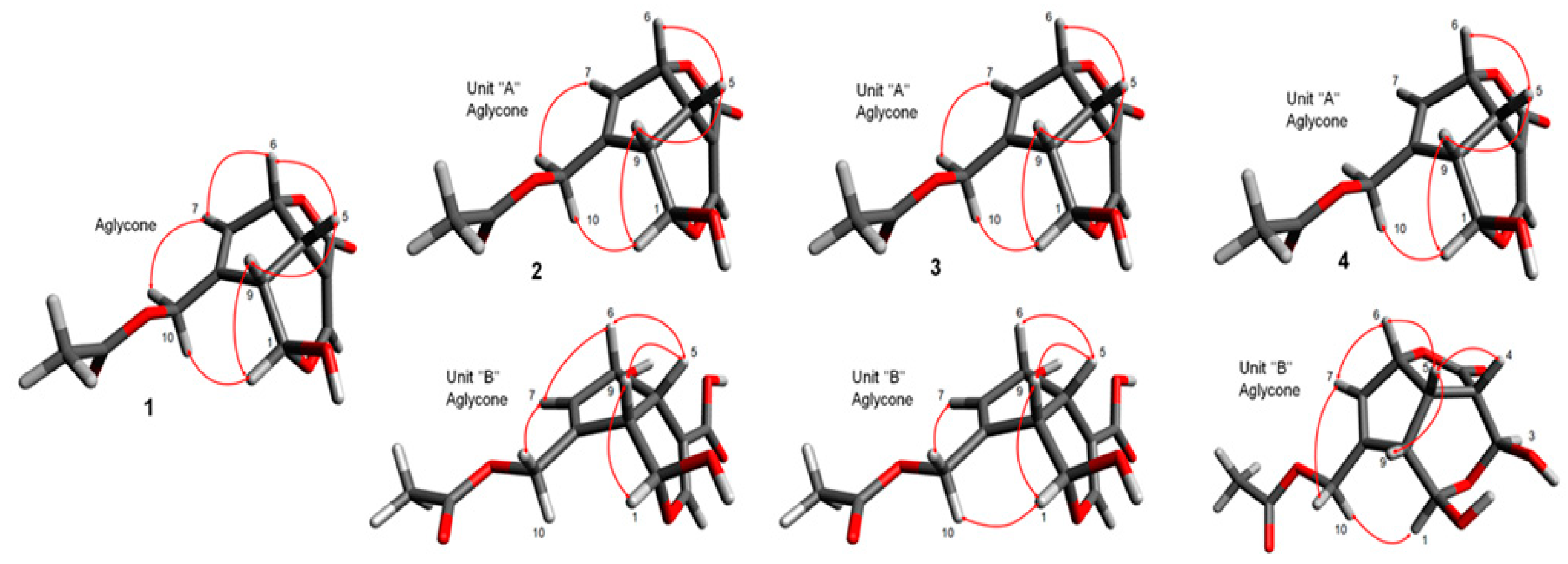
2.1.1. Chemical Structure of Compound 1
2.1.2. Chemical Structure of Compound 2
2.1.3. Chemical Structure of Compound 3
2.1.4. Chemical Structure of Compound 4
3. Materials and Methods
3.1. General Methods
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Spectroscopic Data of Compounds 1–4
3.5. Acid Hydrolysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Robbrecht, E.; Manen, J.F. The major evolutionary lineages of the coffee family (Rubiaceae, angiosperms). Combined analysis (nDNA and cpDNA) to infer the position of Coptosapelta and Luculia, and supertree construction based on rbcL, rps16, trnLtrnF and atpB-rbcL data. A new classification in two subfamilies, Cinchonoideae and Rubioideae. Syst. Geogr. Plants 2006, 76, 85–146. [Google Scholar]
- Briggs, L.H.; Nicholls, G.A. Chemistry of the coprosma genus VIII. The occurrence of asperuloside. J. Chem. Soc. 1954, 1954, 3940–3943. [Google Scholar] [CrossRef]
- Kooiman, P. The occurrence of asperulosidic glycosides in the Rubiaceae. Acta Bot. Neerl. 1969, 18, 124–137. [Google Scholar] [CrossRef]
- Inouye, H.; Takeda, Y.; Nishimura, H.; KANOMI, A.; Okuda, T.; Puff, C. Chemotaxonomic studies of Rubiaceous plant containing iridoid glycosides. Phytochemistry 1988, 27, 2591–2598. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, C.; Liu, X.; Zhang, Y.; Wang, K.; Ceng, Z. Chemical constituents and antioxidant activity of Lasianthus hartii. Chem. Nat. Compd. 2017, 53, 390–393. [Google Scholar] [CrossRef]
- Li, B.; Lai, X.W.; Xu, X.H.; Yu, B.W.; Zhu, Y. A new anthraquinone from the root of Lasianthus acuminatissimus. Yao Xue Xue Bao 2007, 42, 502–504. [Google Scholar] [PubMed]
- Li, B.; Zhang, D.M.; Luo, Y.M.; Chen, X. Three new antitumor anthraquinone glycosides from Lasianthus acuminatissimus Merr. Chem. Pharm. Bull. 2006, 54, 297–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhang, D.M.; Luo, Y.M. A new sesquiterpene lactone from roots of Lasianthus acuminatissimus. Yao Xue Xue Bao 2006, 41, 426–430. [Google Scholar] [PubMed]
- Li, B.; Zhang, D.M.; Luo, Y.M. Chemical constituents from root of Lasianthus acuminatissimus I. Zhongguo Zhong Yao Za Zhi 2006, 31, 133–135. [Google Scholar] [PubMed]
- Dallavalle, S.; Jayasinghe, L.; Kumarihamy, B.M.M.; Merlini, L.; Musso, L.; Scaglioni, L. A new 3,4-seco-lupane derivative from Lasianthus gardneri. J. Nat. Prod. 2004, 67, 911–913. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Shimidzu, H.; Mizuno, K.; Inouchi, S.; Masuda, T.; Hirata, E.; Shinzato, T.; Aramoto, M.; Otsuka, H. An iridoid glucoside dimer and a non-glycosidic iridoid from the leaves of Lasianthus wallichii. Chem. Pharm. Bull. 2002, 50, 1395–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hamoud, G.A.; Orfali, R.S.; Perveen, S.; Mizuno, K.; Takeda, Y.; Nehira, T.; Masuda, K.; Sugimoto, S.; Yamano, Y.; Otsuka, H.; et al. Lasianosides A–E: New iridoid glucosides from the leaves of Lasianthus verticillatus (Lour.) Merr. and their antioxidant activity. Molecules 2019, 24, 3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, H.; Yoshimura, K.; Yamasaki, K.; Cantoria, M.C. Isolation of 10-O-Acyl Iridoid glucosides from a Philippine medicinal plant, Oldenlandia corymbose L. (Rubiaceae). Chem. Pharm. Bull. 1991, 39, 2049–2052. [Google Scholar] [CrossRef] [Green Version]
- Taskova, R.M.; Gotfredsen, C.H.; Jensen, S.R. Chemotaxonomy of Veroniceae and its allies in the Plantaginaceae. Phytochemistry 2006, 67, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Podanyi, B.; Kocsis, A.; Szabo, L.; Reid, R.S. An NMR study of the solution conformation of two asperuloside derivatives. Phytochemistry 1990, 29, 861–866. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1–8 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δc | δH Multi (J in Hz) |
|---|---|---|
| 1 | 93.3 | 5.98 d (1.0) |
| 3 | 150.4 | 7.32 d (2.0) |
| 4 | 106.2 | - |
| 5 | 37.5 | 3.70 t-like (6.8) |
| 6 | 86.4 | 5.59 br dt (6.5, 1.7) |
| 7 | 129.1 | 5.75 br t (1.8) |
| 8 | 144.4 | - |
| 9 | 45.4 | 3.31 m |
| 10 | 61.7 | 4.69 dd (14.4, 1.0) 4.81 dd (14.4, 1.0) |
| 11 | 172.6 | - |
| 1’ | 100.0 | 4.70 d (7.9) |
| 2’ | 74.7 | 3.22 dd (9.1, 7.9) |
| 3’ | 77.9 | 3.40 br t (8.9) |
| 4’ | 71.6 | 3.30 m |
| 5’ | 78.4 | 3.37 ddd (9.3, 6.0,2.1) |
| 6’ | 62.8 | 3.70 dd (11.8, 6.0) 3.94 dd (11.8, 2.1) |
| 1’’ | 174.2 | - |
| 2’’ | 44.0 | 2.28, 2H, d (7.2) |
| 3’’ | 26.8 | 2.09 nonet-like (6.8) |
| 4’’/5’’ | 22.8 | 0.98, 6H, d (6.6) |
| Position | 2 | 3 | ||
|---|---|---|---|---|
| Unit A δH Multi (J in Hz) | Unit B δH Multi (J in Hz) | Unit A δH Multi (J in Hz) | Unit B δH Multi (J in Hz) | |
| 1 | 5.86 d (1.4) | 5.05 d (9.1) | 5.99 br s | 5.07 d (9.2) |
| 3 | 7.15 d (1.9) | 7.70 d (1.1) | 7.33 br d (1.7) | 7.77 br d (1.3) |
| 4 | - | - | - | - |
| 5 | 3.43 m | 2.86 ddd (7.1, 5.7, 1.1) | 3.69 m | 3.12 br t (6.8) |
| 6 | 5.51 br dt (6.6, 1.6) | 4.80 m | 5.59 br t (6.2) | 4.89 m |
| 7 | 5.69 br t (1.6) | 6.00 br d (1.7) | 5.76 br s | 6.05 br d (1.9) |
| 8 | - | - | - | - |
| 9 | 3.27 m | 2.73 t-like (8.5) | 3.36 m | 2.72 t-like (8.1) |
| 10 | 4.62 dd (14.6, 0.9) 4.74 m | 4.82 m 4.95 dd (14.8, 0.6) | 4.69 br d (14.4) 4.80 m | 4.82 m 4.97 br d (15.8) |
| 11 | - | - | - | - |
| 10-COCH3 | - | - | - | - |
| 10-COCH3 | 2.05 s | 2.09 s | 2.10 s | 2.10 s |
| 1’ | 4.92 d (8.2) | 4.70 d (7.8) | 4.84 m | 4.75 d (7.9) |
| 2’ | 4.80 m | 3.27 m | 3.44 dd (9.6, 8.1) | 3.27 t-like (8.3) |
| 3’ | 3.67 t-like (9.2) | 3.38 m | 5.08 t-like (8.6) | 3.30 m |
| 4’ | 3.38 m | 3.29 m | 3.59 t-like (9.4) | 3.28 m |
| 5’ | 3.45 m | 3.29 m | 3.51 ddd (9.8, 5.6, 1.9) | 3.40 br d (8.8) |
| 6’ | 3.69 dd (11.9, 6.7) 3.94 dd (11.9, 1.8) | 3.61 dd (12.0, 5.6) 3.83 dd (12.0, 1.4) | 3.74 m 3.95 dd (11.9, 1.9) | 3.63 dd (11.9, 5.8) 3.87 dd (11.9, 1.7) |
| Position | 2 | 3 * | ||
|---|---|---|---|---|
| Unit A | Unit B | Unit A | Unit B | |
| 1 | 94.1 | 101.8 | 93.4 | 101.4 |
| 3 | 150.1 | 156.3 | 150.3 | 156.2 |
| 4 | 106.3 | 107.7 | 106.2 | 108.0 |
| 5 | 37.6 | 42.9 | 37.5 | 42.5 |
| 6 | 86.1 | 75.2 | 86.4 | 75.8 |
| 7 | 129.1 | 131.6 | 129.0 | 131.5 |
| 8 | 143.9 | 146.5 | 144.3 | 146.2 |
| 9 | 45.1 | 45.9 | 45.3 | 46.4 |
| 10 | 61.9 | 63.8 | 62.0 | 63.8 |
| 11 | 172.2 | 167.6 | 172.6 | 168.6 |
| 10-COCH3 | 172.7 | 172.2 | 172.4 | 172.6 |
| 10-COCH3 | 20.6 | 20.8 | 20.7 | 20.8 |
| 1’ | 98.7 | 100.9 | 100.0 | 100.6 |
| 2’ | 74.5 | 74.9 | 73.0 | 74.9 |
| 3’ | 75.6 | 77.8 | 78.7 | 77.9 |
| 4’ | 71.6 | 71.7 | 70.0 | 71.6 |
| 5’ | 78.5 | 78.6 | 78.6 | 78.6 |
| 6’ | 62.7 | 63.0 | 62.4 | 63.0 |
| Position | Unit A | Unit B | ||
|---|---|---|---|---|
| δc | δH multi (J in Hz) | δc | δH multi (J in Hz) | |
| 1 | 93.5 | 5.91 d (1.1) | 97.0 | 5.14 d (6.0) |
| 3 | 150.3 | 7.33 d (1.9) | 97.4 | 5.27 d (3.6) |
| 4 | 106.3 | - | 44.4 | 3.36 m |
| 5 | 37.5 | 3.69 td-like (6.8, 1.8) | 37.5 | 3.47 m |
| 6 | 86.4 | 5.59 dt (6.6, 1.4) | 87.9 | 5.41 br d (6.5) |
| 7 | 128.9 | 5.75 br s | 125.9 | 6.01 br s |
| 8 | 144.3 | - | 152.5 | - |
| 9 | 45.5 | 3.37 m | 46.3 | 3.05 m |
| 10 | 61.9 | 4.68 br dd (14.3, 1.0) 4.79 dd (14.6, 1.2) | 62.8 | 4.68 dd-like (14.3, 1.0) 5.00 br d (15.9) |
| 11 | 172.6 | - | 176.9 | - |
| 10-COCH3 | 172.2 | - | 172.6 | - |
| 10-COCH3 | 20.8 | 2.13 s | 20.8 | 2.09 s |
| 1’ | 100.2 | 4.75 d (8.1) | 99.6 | 4.73 d (8.1) |
| 2’ | 74.5 | 3.24 dd (8.9, 8.1) | 75.0 | 3.24 dd (8.9, 8.1) |
| 3’ | 77.8 | 3.42 m | 77.9 | 3.43 m |
| 4’ | 71.2 | 3.42 m | 71.6 | 3.30 m |
| 5’ | 76.6 | 3.57 br dd (9.4, 3.6) | 78.4 | 3.32 m |
| 6’ | 68.1 | 3.95 dd (11.7, 1.4) 4.18 dd (11.7, 5.0) | 62.8 | 3.67 dd (11.7, 3.6) 3.88 br d (11.7) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Hamoud, G.A.; Orfali, R.S.; Takeda, Y.; Sugimoto, S.; Yamano, Y.; Al Musayeib, N.M.; Fantoukh, O.I.; Amina, M.; Otsuka, H.; Matsunami, K. Lasianosides F–I: A New Iridoid and Three New Bis-Iridoid Glycosides from the Leaves of Lasianthus verticillatus (Lour.) Merr. Molecules 2020, 25, 2798. https://doi.org/10.3390/molecules25122798
Al-Hamoud GA, Orfali RS, Takeda Y, Sugimoto S, Yamano Y, Al Musayeib NM, Fantoukh OI, Amina M, Otsuka H, Matsunami K. Lasianosides F–I: A New Iridoid and Three New Bis-Iridoid Glycosides from the Leaves of Lasianthus verticillatus (Lour.) Merr. Molecules. 2020; 25(12):2798. https://doi.org/10.3390/molecules25122798
Chicago/Turabian StyleAl-Hamoud, Gadah Abdulaziz, Raha Saud Orfali, Yoshio Takeda, Sachiko Sugimoto, Yoshi Yamano, Nawal M. Al Musayeib, Omer Ibrahim Fantoukh, Musarat Amina, Hideaki Otsuka, and Katsuyoshi Matsunami. 2020. "Lasianosides F–I: A New Iridoid and Three New Bis-Iridoid Glycosides from the Leaves of Lasianthus verticillatus (Lour.) Merr." Molecules 25, no. 12: 2798. https://doi.org/10.3390/molecules25122798