Antiparasitic Constituents of Beilschmiedia louisii and Beilschmiedia obscura and Some Semisynthetic Derivatives (Lauraceae)

 , ,
, ,
Abstract
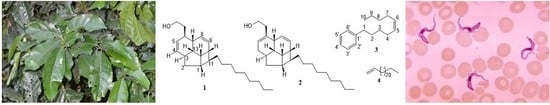
:
1. Introduction
2. Results and Discussion
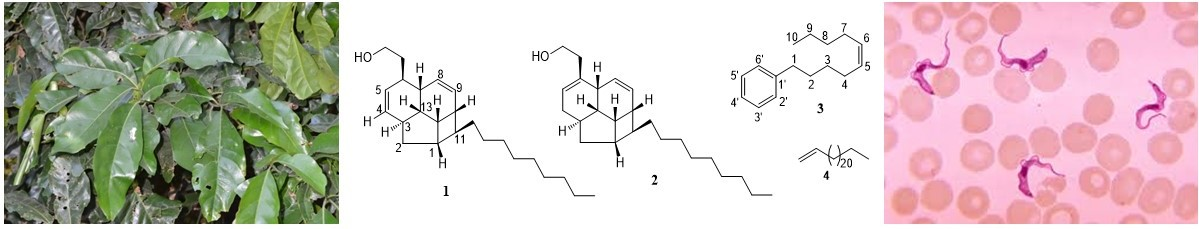
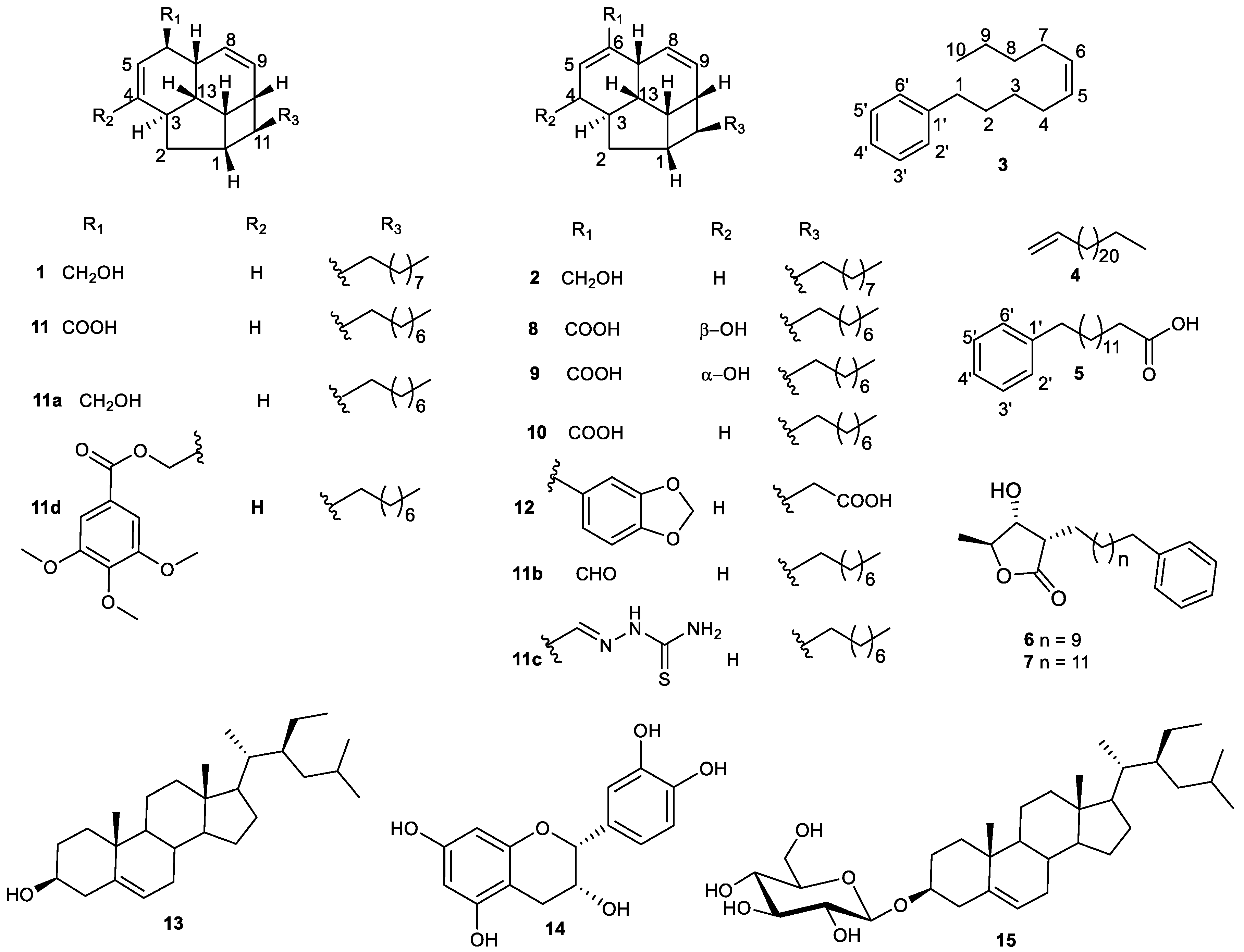
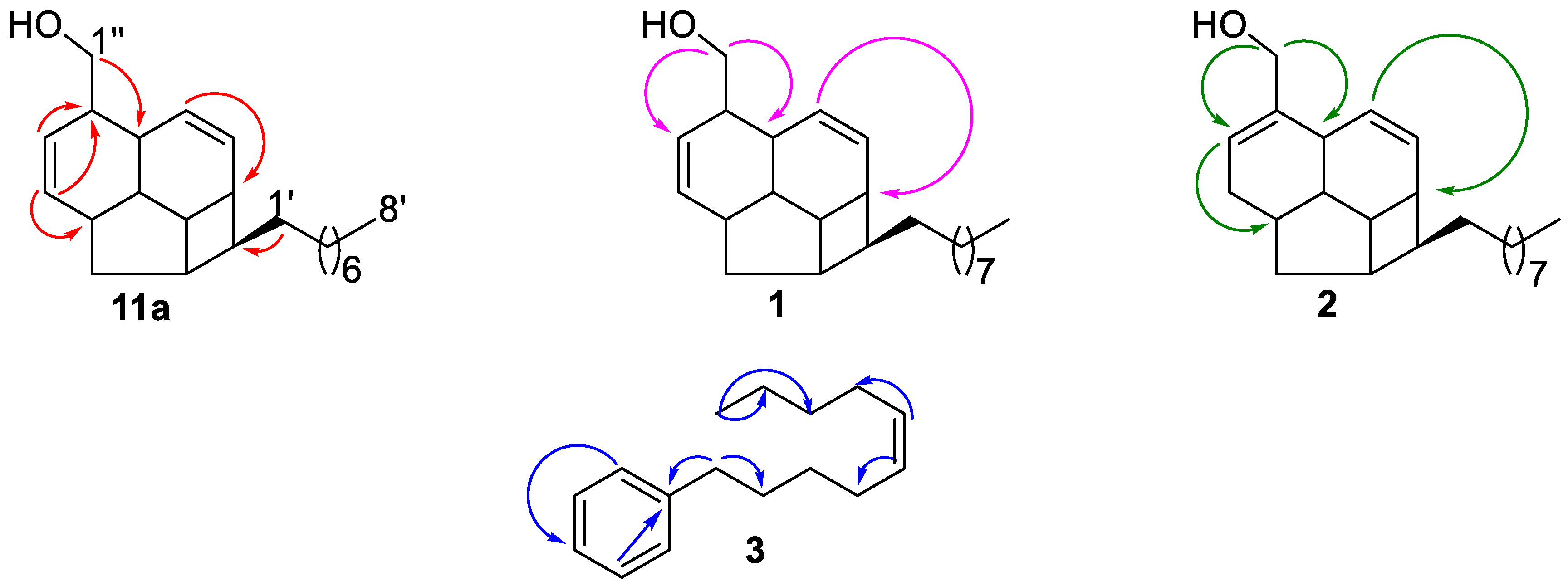
2.1. Structure Elucidation
2.2. In Vitro Antiplasmodial and Antitrypanosomal Activities
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instruments and Apparatus
3.3. Plant Material
3.4. Extraction and Isolation
3.5. Semi-Synthetic Reactions
3.5.1. Synthesis of Compound 11a (Beilschmiedol A)
3.5.2. Synthesis of Compound 11b (Beilschmiedal)
3.5.3. Synthesis of Compound 11c (Beilschmiecarbazone)
3.5.4. Synthesis of Compound 11d (Beilschmiegallate)
3.6. Spectroscopic Data
3.7. Biological Activities
3.7.1. In Vitro Antiplasmodial Assay
3.7.2. In Vitro Antitrypanosomal Assay
3.7.3. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fouilloy, R. Lauraceae. In Flore du Cameroun; Muséum National d’Histoire Naturelle: Paris, France, 1974; Volume 18, pp. 3–87. [Google Scholar]
- Liao, C. Lauraceae. In Flora of Taiwan; Editorial Committee of the flora of Taiwan: Tapei, Tawain, 1996; Volume 2, pp. 433–499. [Google Scholar]
- Lenta, B.N.; Chouna, J.R.; Nkeng-Efouet, P.A.; Sewald, N. Endiandric acid derivatives and other constituents of plants from the genera Beilschmiedia and Endiandra (Lauraceae). Biomolecules 2015, 5, 910–942. [Google Scholar] [CrossRef] [Green Version]
- Iwu, M.M. Handbook of African Medicinal Plants; CRC Press, 2000 Corporate Blvd., N.W.: Boca Raton, FL, USA, 1993; p. 435. [Google Scholar]
- Robyns, W.; Staner, P.; Demaret, F.; Germain, R.; Gilbert, G.; Hauman, L.; Homès, M.; Jurion, F.; Lebrun, J.; Abeele, V. Flore du Congo Belge et du Ruanda-Urundi. Spermatophytes; Institut National pour l’Étude Agronomique du Congo Belge: Bruxelles, Belgique, 1952; Volume 2, pp. 403–446. [Google Scholar]
- Bolza, E.; Keating, W.G. African Timbers: The Properties, Uses and Characteristics of 700 Species; Division of Building Research, CSIRO: Melbourne, Australia, 1972; p. 710. [Google Scholar]
- Apel, C.; Geny, C.; Dumontet, V.; Birlirakis, N.; Roussi, F.; Pham, V.C.; Doan Thi Mai, H.; Nguyen, V.H.; Chau, V.M.; Litaudon, M. Endiandric acid analogues from Beilschmiedia ferruginea as dual inhibitors of Bcl-xL/Bak and Mcl-1/Bid interactions. J. Nat. Prod. 2014, 77, 1430–1437. [Google Scholar] [CrossRef]
- Lenta, B.N.; Chouna, J.R.; Nkeng-Efouet, P.A.; Kimbu, S.F.; Tsamo, E.; Sewald, N. A new cyclostachine acid derivative from Beilschmiedia obscura. Nat. Prod. Commun. 2011, 6, 1591–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesołowska, A.; Grzeszczuk, M.; Kulpa, D. GC-MS analysis of the essential oil from flowers of Chrysanthemum coronarium L. propagated conventionally and derived from in vitro cultures. Acta Chromatogr. 2015, 3, 525–539. [Google Scholar] [CrossRef]
- Pinto, J.; Oliveira, A.S.; Lopes, P.; Roseira, I.; Cabral, M.; de Lourdes Bastos, M.; Guedes de Pinho, P. Characterization of chemical compounds susceptible to be extracted from cork by the wine using GC-MS and 1H–NMR metabolomic approaches. Food Chem. 2019, 271, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Pupo, M.T.; Vieira, P.C.; Fernandes, J.B.; Fátima, M.; da Silva, M.F. A cycloartane triterpenoid and to-phenyl alkanoic and alkenoic acids from Trichilia claussenii. Phytochemistry 1996, 42, 795–798. [Google Scholar] [CrossRef]
- Pupo, M.T.; Vieira, P.C.; Fernandes, J.B.; Fátima, M.; da Silva, G.F. γ-Lactones from Trichilia claussenii. Phytochemistry 1998, 48, 307–310. [Google Scholar] [CrossRef]
- Chouna, J.R.; Nkeng-Efouet, P.A.; Lenta, B.N.; Devkota, K.P.; Neumann, B.; Stammler, H.-G.; Kimbu, S.F.; Sewald, N. Antibacterial endiandric acid derivatives from Beilschmiedia anacardioides. Phytochemistry 2009, 70, 684–688. [Google Scholar] [CrossRef]
- Chouna, J.R.; Nkeng-Efouet, P.A.; Lenta, B.N.; Wansi, J.D.; Kimbu, S.F.; Sewald, N. Endiandric acid derivatives from the stem bark of Beilschmiedia anacardioides. Phytochem. Lett. 2010, 3, 13–16. [Google Scholar] [CrossRef]
- Banfield, J.E.; Black, D.S.; Collins, D.J.; Hyland, B.P.M.; Lee, J.J.; Pranowo, S.R. Constituents of some species of Beilschmiedia and Endiandra (Lauraceae): New endiandric acid and benzopyran derivatives isolated from B. oligandra. Aust. J. Chem. 1994, 47, 587–607. [Google Scholar] [CrossRef]
- Chaturvedula, V.S.P.; Prakash, I. Isolation of stigmasterol and β-sitosterol from the dichloromethane extract of Rubussua vissimus. Int. Curr. pharm. 2012, 1, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Ganapaty, S.; Pannakal, S.T.; Srilakshmi, G.V.K.; Lakshmi, P.; Watterman, P.G.; Brun, R. Pumilanol, an antiprotozoal isoflavanol from Tephrosia pumila. Phytochem. Lett. 2008, 1, 175–178. [Google Scholar] [CrossRef]
- Faizi, S.; Ali, M.; Saleem, R.; Irfanullah; Bibi, S. Complete 1H and 13C–NMR assignments of stigma-5-en-3-O-β-glucoside and its acetyl derivative. Magn. Reson. Chem. 2001, 39, 399–405. [Google Scholar] [CrossRef]
- Chen, X.; Klöckner, J.; Holze, J.; Zimmermann, C.; Seemann, W.K.; Schrage, R.; Bock, A.; Mohr, K.; Tränkle, C.; Holzgrabe, U.; et al. Rational design of partial agonists for the muscarinic M1 acetylcholine receptor. J. Med. Chem. 2015, 58, 560–576. [Google Scholar] [CrossRef]
- Bandaranayake, W.M.; Banfield, J.E.; Black, D.S.C.; Fallon, G.D.; Gatehouse, B.M. Endiandric acid, a novel carboxylic acid from Endiandra introrsa (Lauraceae): X-ray structure determination. J. Chem. Soc. Chem. Commun. 1980, 4, 162–163. [Google Scholar] [CrossRef]
- Bandaranayake, W.M.; Banfield, J.E.; Black, D.S.C.; Fallon, G.D.; Gatehouse, B.M. Constituents of Endiandra species. Endiandric acid, a novel carboxylic acid from Endiandra introrsa Lauraceae), and a derived lactone. Aust. J. Chem. 1981, 34, 1655–1667. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar] [CrossRef]
- Fatondji, H.R.; Gbaguidi, F.; Kpoviessi, S.; Bero, J.; Hannaert, V.; Quetin-Leclercq, J.; Poupaert, J.; Moudachirou, M.; Accrombessi, G.C. Synthesis, characterization and trypanocidal activity of some aromatic thiosemicarbazones and their 1,3,4 thiadiazolines derivatives. Afr. J. Pure Appl. Chem. 2011, 5, 59–64. [Google Scholar]
- Uwe, J.; Mundinger, S.; Bannwarth, W. Efficient transfer of chelating amides into different types of esters and lactones. Eur. J. Org. Chem. 2014, 2014, 6963–6974. [Google Scholar]
- Nicolaou, K.C.; Petasis, N.A.; Zipkin, R.E.; Uenishi, J. The endiandric acid cascade. Electrocyclizations in organic synthesis. I. Stepwise, stereocontrolled total synthesis of endiandric acids A and B. J. Am. Chem. Soc. 1982, 104, 5555–5557. [Google Scholar] [CrossRef]
- Bankeu, J.J.K.; Dawé, A.; Mbiantcha, M.; Feuya, G.R.T.; Ali, I.; Tchuenmogne, M.A.T.; Mehreen, L.; Lenta, B.N.; Ali, M.S.; Ngouela, A.S. Characterization of bioactive compounds from Ficus vallis-choudae Delile (Moraceae). Trends Phytochem. Res. 2017, 1, 235–242. [Google Scholar]
- Bankeu, J.J.K.; Mustafa, S.A.A.; Gojayev, A.S.; Lenta, B.D.; Noungoué, D.T.; Ngouela, A.S.; Asaad, K.; Choudhary, M.I.; Prigge, S.; Guliyev, A.A.; et al. Ceramide and cerebroside from the stem bark of Ficus mucuso (Moraceae). Chem. Pharm. Bull. 2010, 14, 1661–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Franco, T.; Epenoy, A.; Hu, X. Synthesis of E-alkyl alkenes from terminal alkynes via Ni-catalyzed cross-coupling of alkyl halides with B-alkenyl-9- borabicyclo [3.3.1] nonanes. Org. Lett. 2015, 17, 4910–4913. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, E.A.; Brito, I.A.; Lima, M.L.; Romanelli, M.; Moreira-Filho, J.T.; Neves, B.J.; Andrade, C.H.; Sartorelli, P.; Tempone, A.G.; Costa-Silva, T.A.; et al. Antitrypanosomal activity of acetogenins isolated from the seeds of Porcelia macrocarpa is associated with alterations in both plasma membrane electric potential and mitochondrial membrane potential. J. Nat. Prod. 2019, 82, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.N.H.W.S.; Wan, S.; Farediah, A.; Khong, H.Y.; Razauden, M.Z. A Review on Chemical Constituents and Biological Activities of the Genus Beilschmiedia (Lauraceae). Trop. J. Pharm. Res. 2015, 14, 2139–2150. [Google Scholar]
- Pudjastuti, P.; Mukhtar, M.R.; Hadi, A.H.A.; Saidi, N.; Morita, H.; Litaudon, M.; Awang, K. (6,7-Dimethoxy-4-methylisoquinolinyl)-(4′-methoxyphenyl)-methanone, a new benzylisoquinoline alkaloid from Beilschmiedia brevipes. Molecules 2010, 15, 2339–2346. [Google Scholar] [CrossRef]
- Makler, M.T.; Ries, J.M.; Williams, J.A.; Bancroft, J.E.; Piper, R.C.; Gibbins, B.L.; Hinrichs, D.J. Parasite lactate dehydrogenase as an assay for Plasmodium falciparum drug sensitivity. Am. J. Trop. Med. Hyg. 1993, 48, 739–741. [Google Scholar] [CrossRef]
- Hirumi, H.; Martin, S.; Hirumi, K.; Inoue, N.; Kanbara, H.; Saito, A.; Suzuki, N. Cultivation of bloodstream forms of Trypanosoma brucei and T. evansi in a serum-free medium. Trop. Med. Int. Health 1997, 2, 240–244. [Google Scholar] [CrossRef]
- Keusch, G.T.; Jacewicz, M.; Hirschman, S.Z. Quantitative microassay in cell culture for enterotoxin of Shigella dysenteriae 1. J. Infect. Dis. 1972, 125, 539–541. [Google Scholar] [CrossRef]
Sample Availability: Samples of some compounds (4, 8 + 9, 11, 11a, 11c, 13 and 15) are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 11a | |||
|---|---|---|---|---|---|---|
| δC | δH (m, J In Hz) | δC | δH (J In Hz) | δC | δH (J In Hz) | |
| 1 | 41.6 | 2.19, m | 41.4 | 2.18, m | 41.6 | 2.21, m |
| 2a | 35.1 | 1.22, m | 37.4 | 1.40, m | 35.1 | 1.23, m |
| 2b | 1.50, m | 2.54, m | 1.52, m | |||
| 3 | 37.4 | 1.40, m | 36.8 | 1.62, m | 37.4 | 1.43, m |
| 4a | 134.2 | 6.10 (d, 8.0) | 32.1 | 1.82, m | 133.9 | 6.10 (dt, 10.0, 2.4) |
| 4b | 2.18, m | |||||
| 5 | 127.9 | 5.53 (d, 8.0) | 126.0 | 5.74, brs | 127.9 | 5.50 (dt, 10.0, 2.8) |
| 6 | 47.2 | 2.12, m | 141.6 | - | 47.2 | 2.14, m |
| 7 | 35.4 | 2.42, m | 34.4 | 2.67, brs | 35.4 | 2.18, m |
| 8 | 131.3 | 5.47 (d, 8.0) | 126.1 | 5.53, m | 131.4 | 5.39 (brdd, 10.0, 0.8) |
| 9 | 128.9 | 5.48 (brdt, 9.6, 1.8) | 128.2 | 5.53, m | 128.8 | 5.56 (dt, 10.0, 3.2) |
| 10 | 33.3 | 2.53, m | 33.7 | 2.54, s | 33.3 | 2.55, m |
| 11 | 46.3 | 1.40, m | 45.9 | 1.40, m | 46.2 | 1.43, m |
| 12 | 33.1 | 2.42, m | 34.9 | 2.82, m | 32.9 | 2.43, m |
| 13 | 42.7 | 1.50, m | 42.9 | 2.54, m | 42.6 | 1.53, m |
| 1′a | 37.4 | 1.40, m | 37.4 | 1.40, m | 37.4 | 1.43, m |
| 1′b | 2.54, m | 2.54, m | 2.55, m | |||
| 2′ | 27.2 | 1.22, m | 27.2 | 1.22, m | 27.2 | 1.23, m |
| 3′–6′ | 29.5–29.9 | 1.16, m | 29.5–29.9 a | 1.16, m | 29.5–29.9 | 1.23, m |
| 7′ | 32.1 | 1.19, m | 32.1 | 1.16, m | 22.8 | 1.25, m |
| 8′ | 22.8 | 1.22, m | 22.8 | 1.22, m | 14.3 | 0.85 (t, 6.8) |
| 9′ | 14.3 | 0.81 (t, 6.9) | 14.3 | 0.81 (t, 6.9) | ||
| 1″ | 67.6 | 3.63, m | 65.5 | 4.12, m | 67.3 | 3.56, m |
| Position | 11b a | 11c b | 11d a |
|---|---|---|---|
| 1 | 41.4 | 42.2 | 41.4 |
| 2 | 37.2 | 36.8 | 35.0 |
| 3 | 41.9 | 41.5 | 37.4 |
| 4 | 32.1 | 32.8 | 133.9 |
| 5 | 145.9 | 139.5 | 127.1 |
| 6 | 154.0 | 139.7 | 46.1 |
| 7 | 33.3 | 33.1 | 35.2 |
| 8 | 127.8 | 127.8 | 129.0 |
| 9 | 126.1 | 127.6 | 130.5 |
| 10 | 34.6 | 35.0 | 33.2 |
| 11 | 45.9 | 45.9 | 43.6 |
| 12 | 33.4 | 33.8 | 33.2 |
| 13 | 36.8 | 36.8 | 42.4 |
| 1′ | 37.4 | 37.4 | 37.4 |
| 2′ | 27.2 | 27.4 | 27.0 |
| 3′ | 29.8 | 29.9 | 29.7 |
| 4′ | 29.5 | 29.6 | 29.3 |
| 5′ | 29.8 | 29.9 | 29.7 |
| 6′ | 31.3 | 32.1 | 31.9 |
| 7′ | 22.8 | 22.9 | 22.7 |
| 8′ | 14.3 | 14.3 | 14.3 |
| 1″ | 193.9 | 145.5 | 68.9 |
| 1‴ | 180.4 | 166.3 | |
| 2‴ | 125.3 | ||
| 3‴ | 106.8 | ||
| 4‴ | 152.9 | ||
| 5‴ | 142.2 | ||
| 6‴ | 152.9 | ||
| 7‴ | 106.8 | ||
| 4‴/6‴-OMe | 56.2 | ||
| 5‴-OMe | 60.9 |
| Position | 3 | |
|---|---|---|
| δC | δH (J In Hz) | |
| 1 | 36.0 | 2.52, m |
| 2 | 31.6 | 1.53, m |
| 3 | 29.7 | 1.23, brs |
| 4 | 27.3 | 1.98, m |
| 5 | 130.1 | 5.28, m |
| 6 | 130.2 | 5.28, m |
| 7 | 25.7 | 2.69, m |
| 8 | 29.4 | 1.18, brs |
| 9 | 22.6 | 1.21, brs |
| 10 | 14.1 | 0.80 (t, 6.4) |
| 1′ | 142.9 | |
| 2′ | 128.4 | 7.09–7.20, m |
| 3′ | 128.2 | 7.09–7.20, m |
| 4′ | 125.6 | 7.09–7.20, m |
| 5′ | 128.2 | 7.09–7.20, m |
| 6′ | 128.4 | 7.09–7.20, m |
| Position | 4 | |
|---|---|---|
| δC | δH (J In Hz) | |
| 1a | 114.1 | 4.94 (brd, 10.2) |
| 1b | 5.02 (brd, 17.4) | |
| 2 | 139.3 | 5.84, m |
| 3 | 31.9 | 2.07, m |
| 4–23 | 29.4–29.7 | 1.29, brs |
| 24 | 22.7 | 1.39, m |
| 25 | 14.1 | 0.91 (t, 7.2) |
| % Cell Viability ± sd | IC50 a | ||||||
|---|---|---|---|---|---|---|---|
| Sample | Tb Brucei | Pf3D7 | Cytotoxicity | Tb Brucei | Pf3D7 | ||
| B. louisii | Roots | TE | 1.50 ± 0.09 | 99.1 ± 15.3 | 100.0 ± 14.1 | 4.62 | - |
| NF | 19.5 ± 4.2 | 34.8 ± 8.9 | 100.0 ± 1.3 | 20.94 | - | ||
| AF | 79.7 ± 0.6 | 100.0 ± 7.8 | 100.0 ± 19.2 | - | - | ||
| Leaves | TE | 9.7 ± 0.7 | 72.5 ± 3.4 | 100.0 ± 2.3 | 13.82 | - | |
| NF | 97.5 ± 3.5 | 74.6 ± 1.2 | 100.0 ± 4.4 | - | - | ||
| AF | 35.8 ± 10.4 | 100.0 ± 6.2 | 100.0 ± 4.3 | - | - | ||
| B. obscura | Stem bark | TE | 4.4 ± 0.7 | 70.4 ± 2.1 | 100.0 ± 5.5 | 21.59 | - |
| NF | nd | nd | nd | nd | nd | ||
| AF | 6.9 ± 1.7 | 69.5 ± 9.6 | 100.0 ± 1.7 | 16.88 | - | ||
| 1 + 2 | 0.45 ± 0.40 | 97.1 ± 3.6 | 100.0 ± 14.0 | 4.91 | - | ||
| 3 | 99.1 ± 1.2 | 85.8 ± 6.0 | 50.3 ± 8.9 | - | - | ||
| 4 | nd | nd | nd | nd | nd | ||
| 5 | 7.13 ± 3.90 | 78.3 ± 4.7 | 76.6 ± 4.1 | 90.10 | - | ||
| 6 + 7 | 0.1 ± 0.8 | 96.5 ± 0.3 | 100.0 ± 6.6 | ≤15.2 * | - | ||
| 8 + 9 | 78.5 ± 1.2 | 95.0 ± 1.1 | 100.0 ± 0.2 | - | - | ||
| 10 | 35.6 ± 6.9 | 100.0 ± 8.1 | 99.8 ± 4.6 | - | - | ||
| 11 | 99.1 ± 7.4 | 100.0 ± 8.1 | 100.0 ± 5.8 | - | - | ||
| 12 | 79.5 ± 8.4 | 100.0 ± 1.9 | 100.0 ± 10.8 | - | - | ||
| 13 | 0.03 ± 0.06 | 82.2 ± 5.7 | 100.0 ± 11.3 | 9.51 | - | ||
| 14 | nd | nd | nd | nd | nd | ||
| 15 | 95.8 ± 2.5 | 100.0 ± 17.9 | 90.5 ± 5.0 | - | - | ||
| 11a | 8.6 ± 0.8 | 79.7 ± 3.7 | 52.6 ± 1.8 | 64.80 | - | ||
| 11b | 10.8 ± 1.2 | 21.8 ± 8.0 | 79.6 ± 17.1 | 26.53 | 31.42 | ||
| 11c | 96.3 ± 6.2 | 92.6 ± 2.9 | 100.0 ± 2.8 | - | - | ||
| 11d | 100.0 ± 9.3 | 96.6 ± 0.8 | 100.0 ± 3.4 | - | - | ||
| Pentamidine | 0.04 | ||||||
| Chloroquine | 0.034 | ||||||
| Emetine | 0.014 | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Waleguele, C.C.; Mba’ning, B.M.; Awantu, A.F.; Bankeu, J.J.K.; Fongang, Y.S.F.; Ngouela, A.S.; Tsamo, E.; Sewald, N.; Lenta, B.N.; Krause, R.W.M. Antiparasitic Constituents of Beilschmiedia louisii and Beilschmiedia obscura and Some Semisynthetic Derivatives (Lauraceae). Molecules 2020, 25, 2862. https://doi.org/10.3390/molecules25122862
Waleguele CC, Mba’ning BM, Awantu AF, Bankeu JJK, Fongang YSF, Ngouela AS, Tsamo E, Sewald N, Lenta BN, Krause RWM. Antiparasitic Constituents of Beilschmiedia louisii and Beilschmiedia obscura and Some Semisynthetic Derivatives (Lauraceae). Molecules. 2020; 25(12):2862. https://doi.org/10.3390/molecules25122862
Chicago/Turabian StyleWaleguele, Christine C., Brice M. Mba’ning, Angelbert F. Awantu, Jean J. K. Bankeu, Yannick S. F. Fongang, Augustin S. Ngouela, Etienne Tsamo, Norbert Sewald, Bruno N. Lenta, and Rui W. M. Krause. 2020. "Antiparasitic Constituents of Beilschmiedia louisii and Beilschmiedia obscura and Some Semisynthetic Derivatives (Lauraceae)" Molecules 25, no. 12: 2862. https://doi.org/10.3390/molecules25122862