
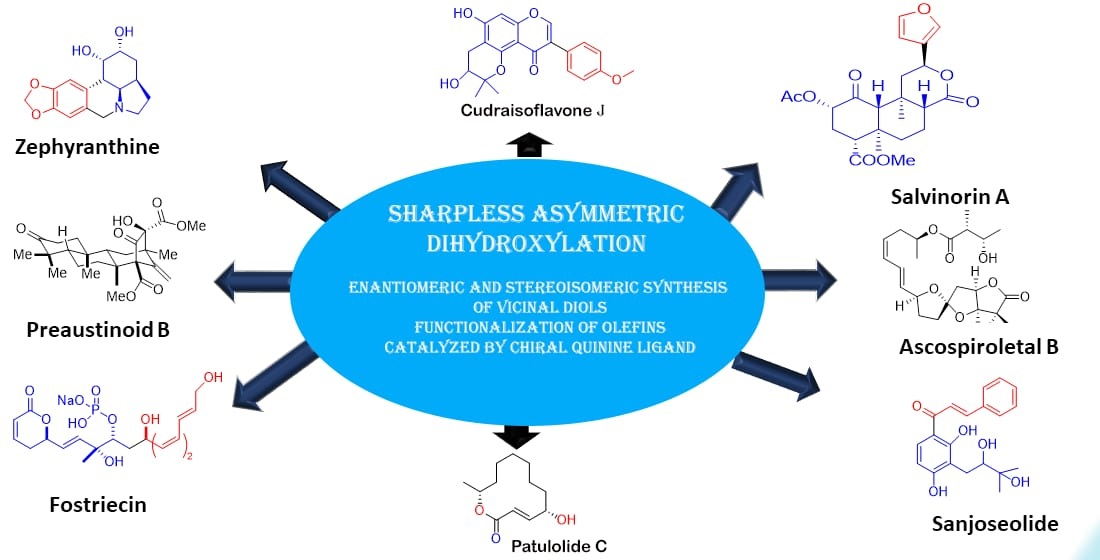
Sharpless Asymmetric Dihydroxylation: An Impressive Gadget for the Synthesis of Natural Products: A Review

 , , ,
, , , 
 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Review of the Literature
2.1. Synthesis of Alkaloid-Based Natural Products
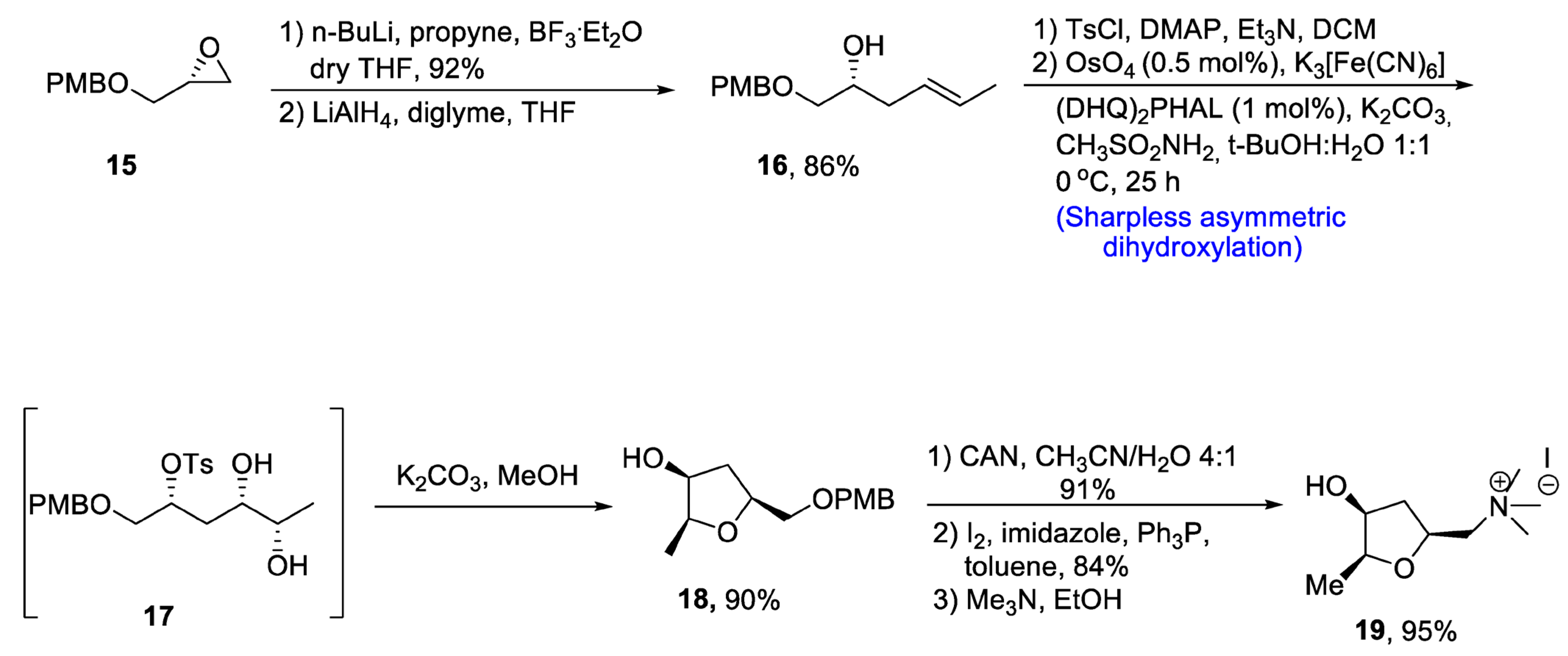
2.1.1. Lycorine-Type Alkaloids
2.1.2. Muscarine Alkaloid
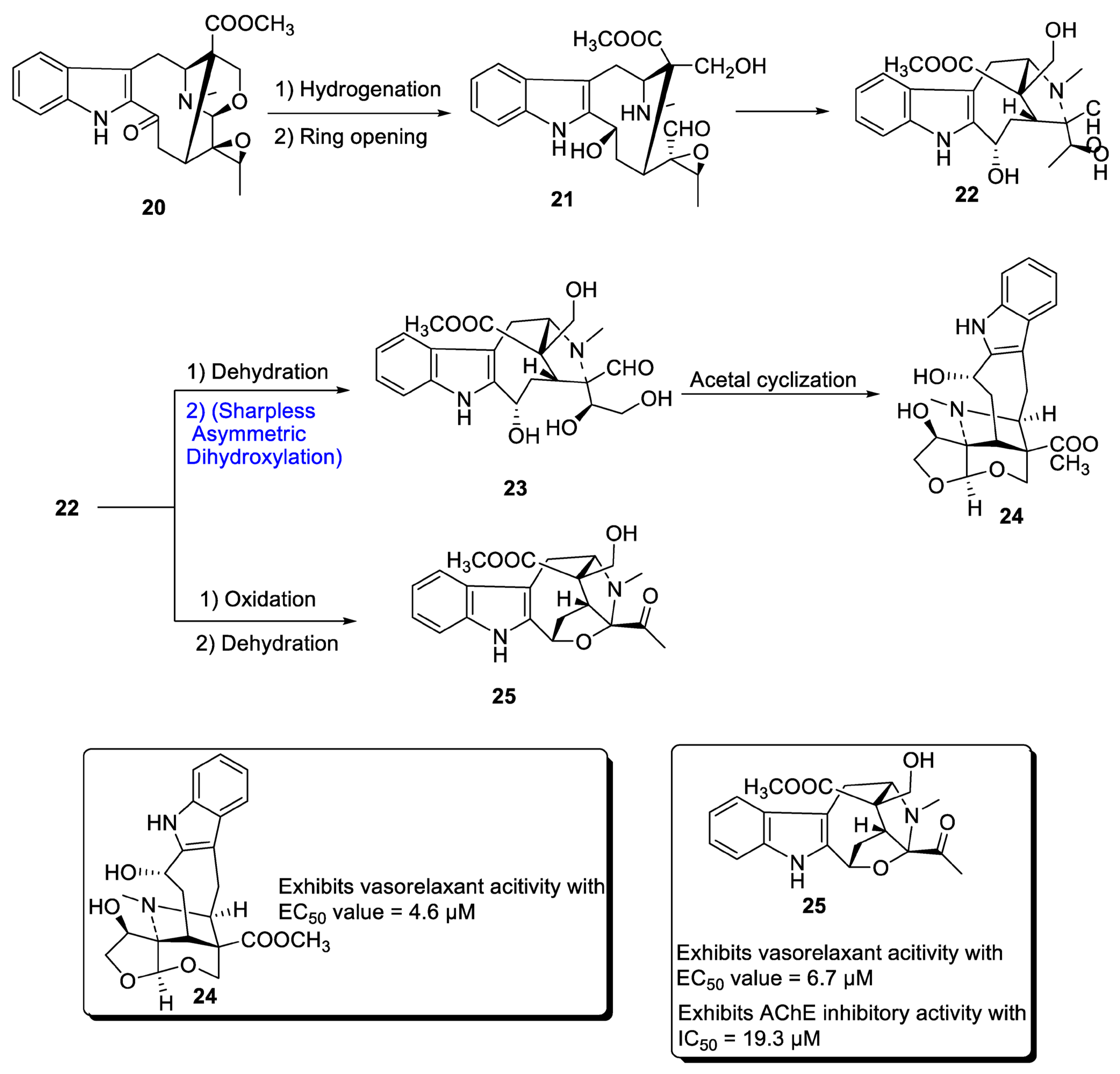
2.1.3. Monoterpenoid Indole Alkaloid
2.1.4. Glyphaeaside Alkaloids
2.2. Synthesis of Terpene-Based Natural Products
2.2.1. Sesquiterpenoids
2.2.2. Neoclerodane Diterpene
2.2.3. Nor-Triterpenoids
2.2.4. Monoterpenoids
2.2.5. Monoterpenoid Alcohol
2.3. Synthesis of Polyketide-Based Natural Products
2.4. Synthesis of Macrolide-Based Natural Products
2.5. Synthesis of Amino Acid-Based Natural Products
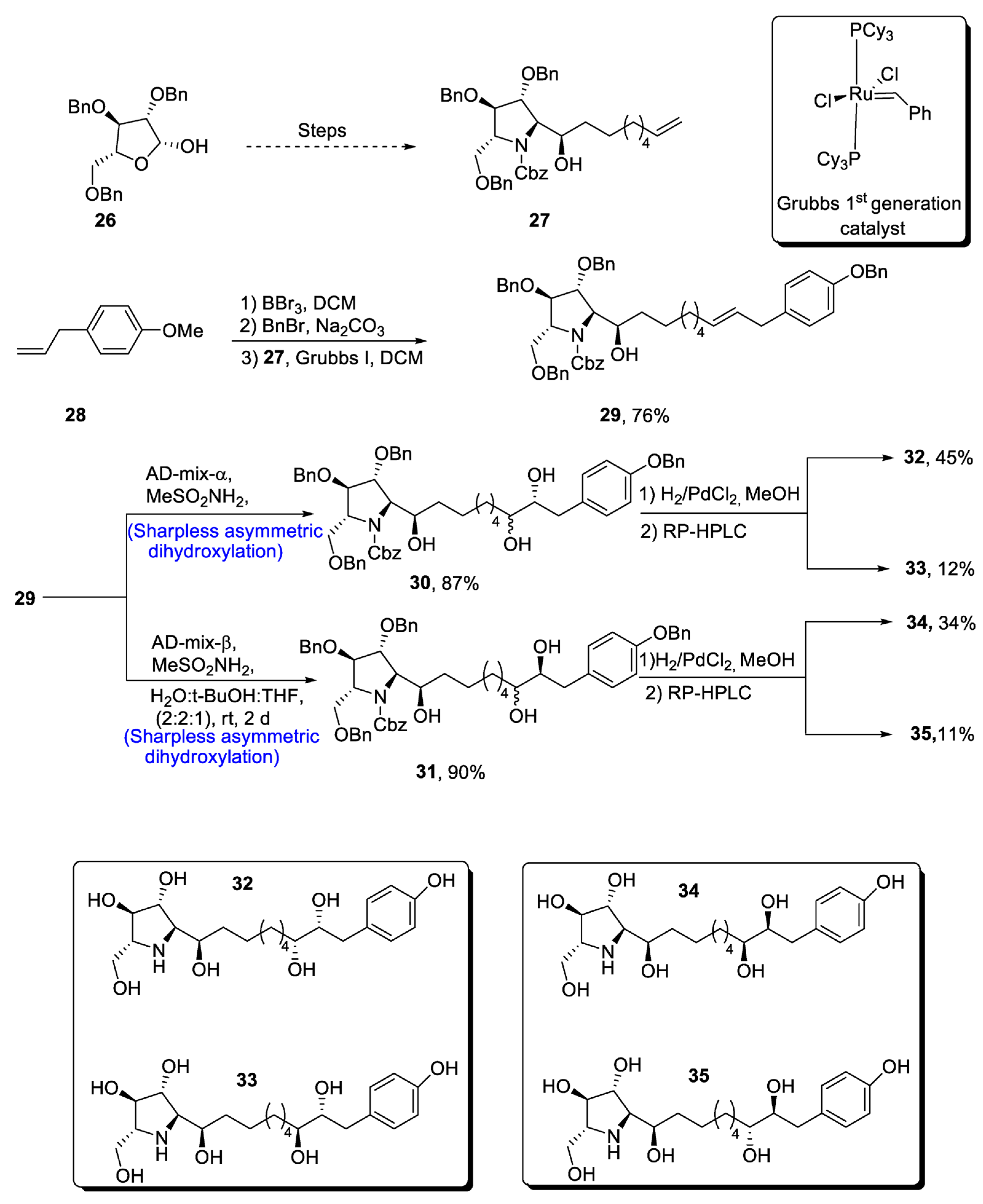
2.5.1. Tubulysins/Amino Acid
2.5.2. Amino Acid Derivative
2.6. Synthesis of Flavonoid-Based Natural Products
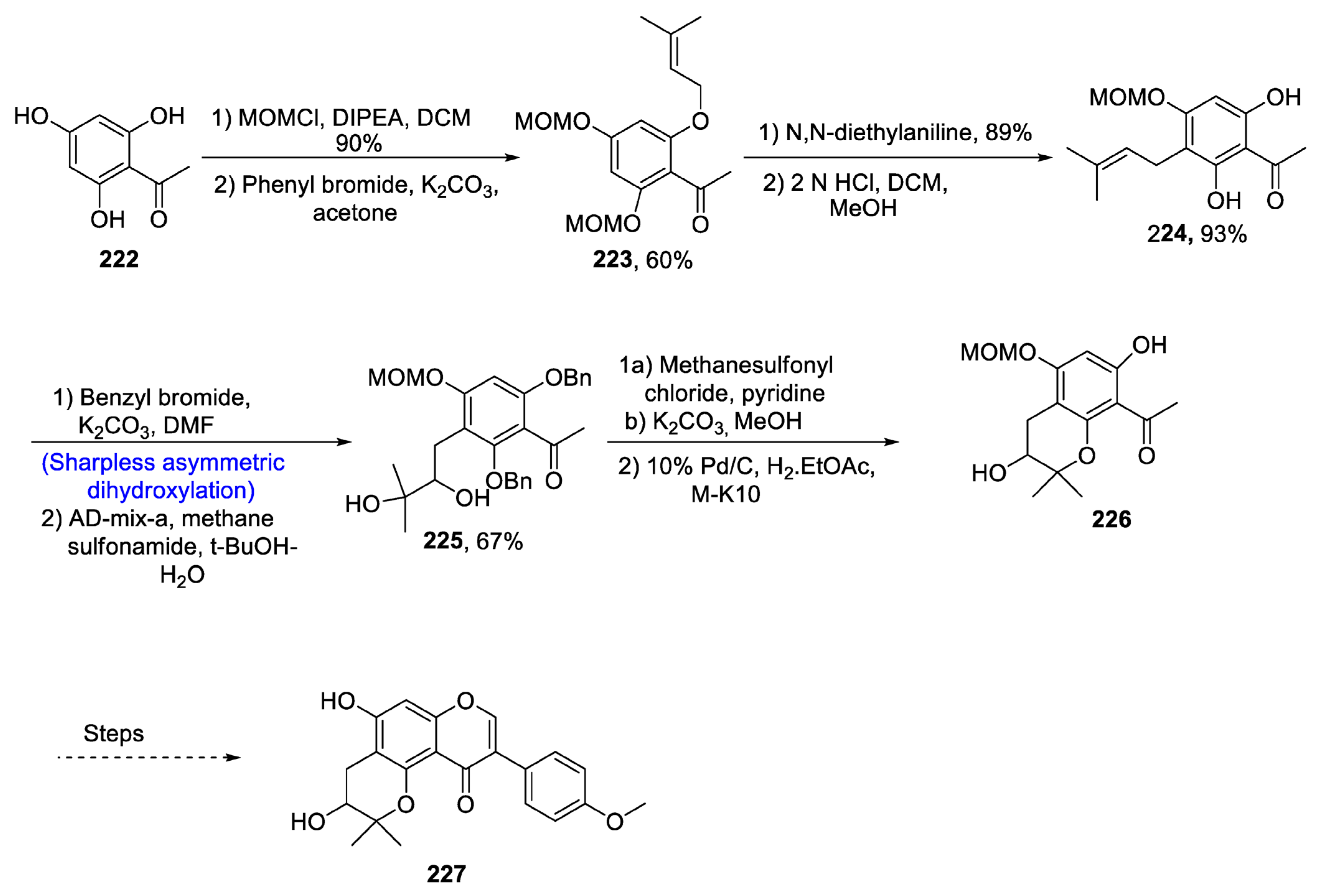
2.6.1. DHPVs/Flavonols
2.6.2. Isoprenylated Chalcone Skeleton/Flavonoid
2.7. Synthesis of Carbohydrate-Based Natural Products
2.8. Synthesis of Lactone-Based Natural Products
2.8.1. Naphthoquinonopyrano-γ-lactone
2.8.2. Containing α,β-Unsaturated Lactone
2.8.3. δ-Lactone Ring Containing Natural Product/ Unsaturated Fatty Acids
2.8.4. Styryllactone
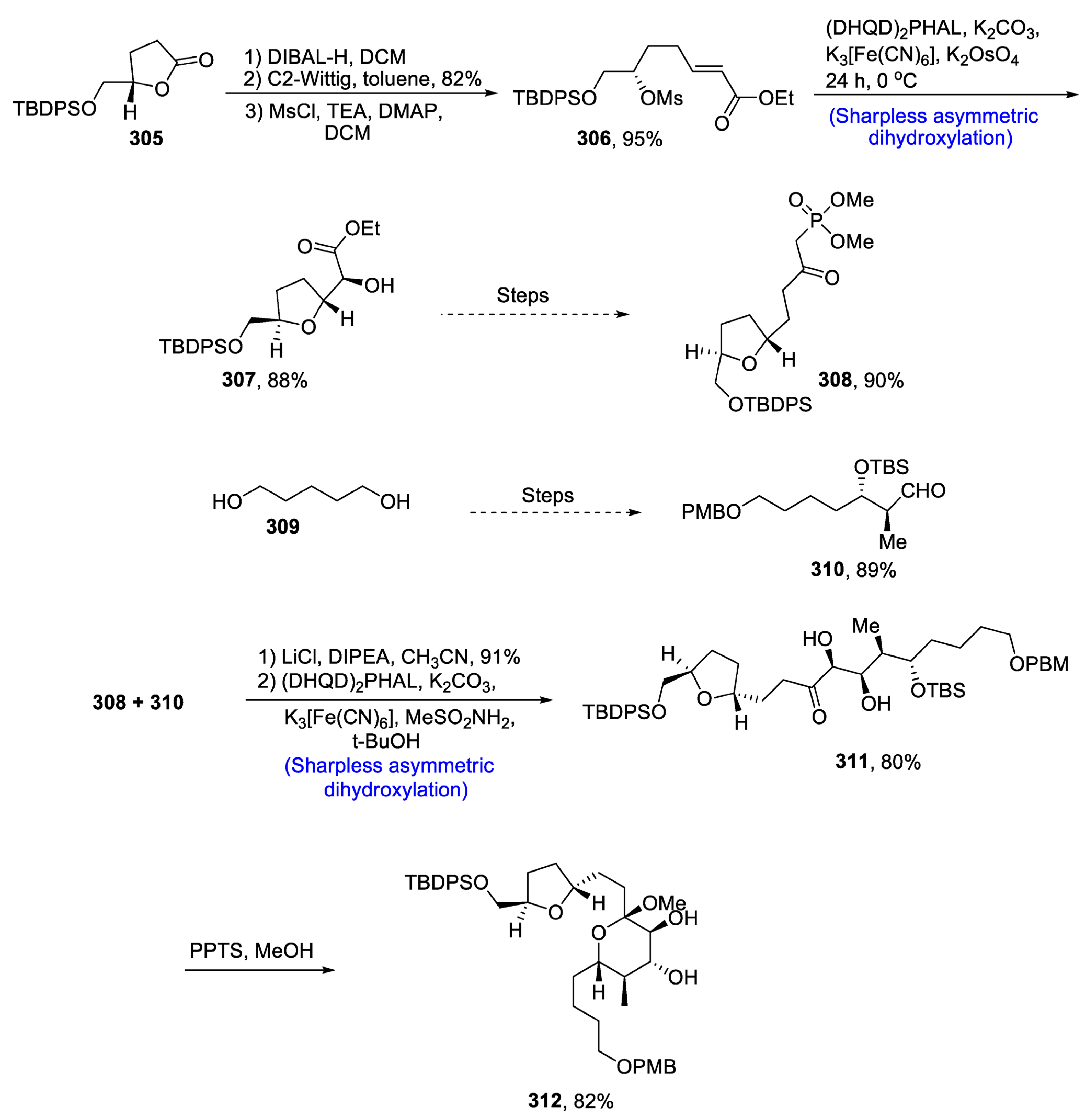
2.9. Synthesis of Tetrahydrofyran Ring-Based Natural Products
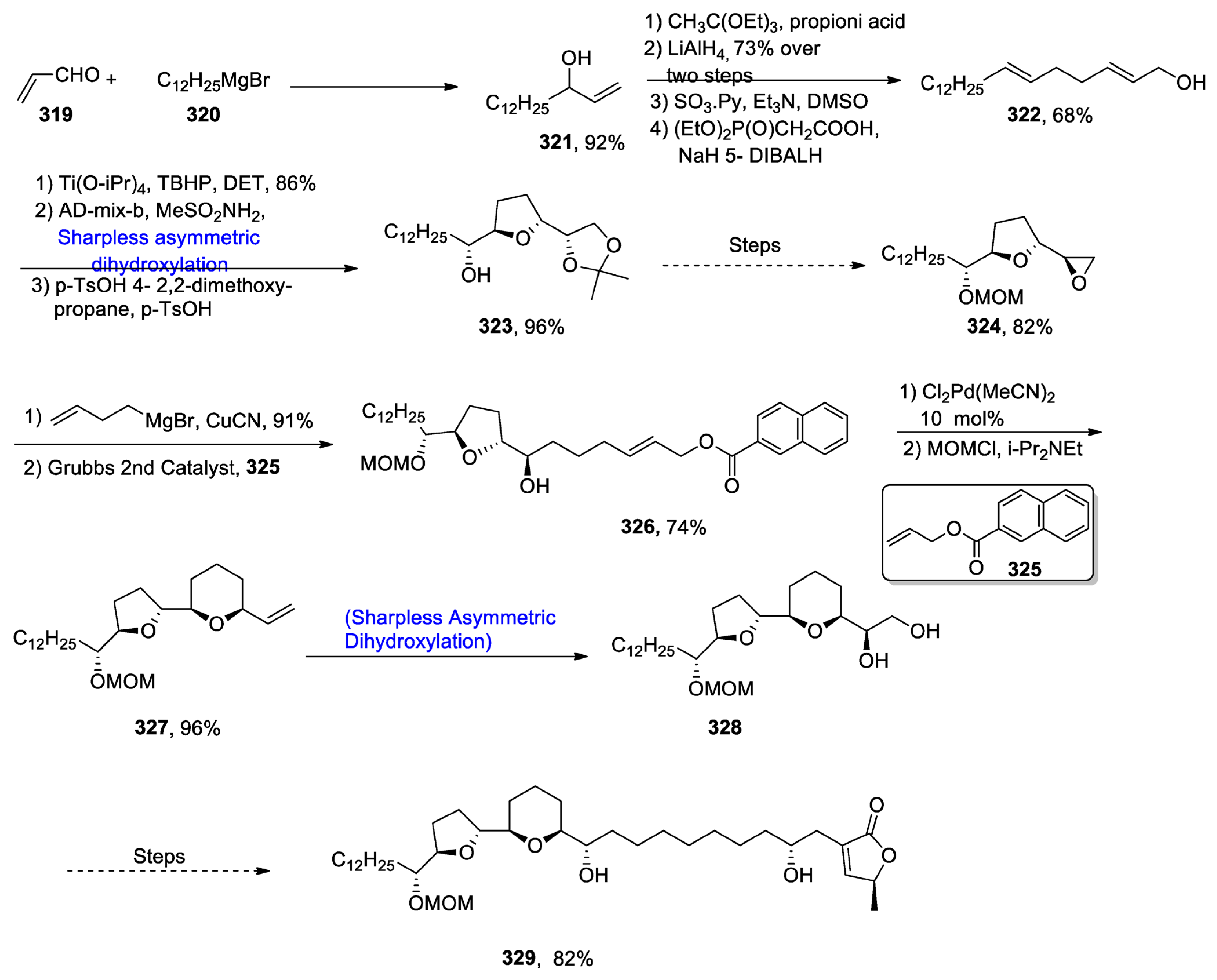
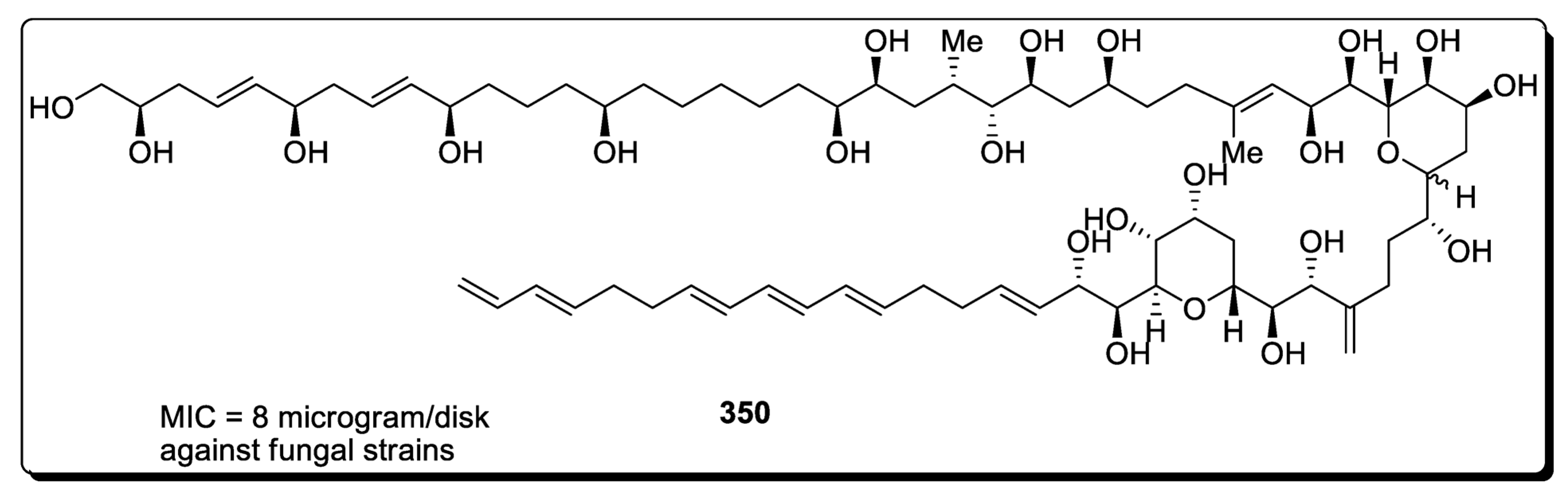
2.10. Miscellaneous Natural Products
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gnas, Y.; Glorius, F. Chiral auxiliaries—Principles and recent applications. Synthesis 2006, 12, 1899–1930. [Google Scholar] [CrossRef]
- Kitano, Y.; Matsumoto, T.; Sato, F. A highly efficient kinetic resolution of γ- and β-trimethylsilyl secondary allylic alcohols by the sharpless asymmetric epoxidation. Tetrahedron 1988, 44, 4073–4086. [Google Scholar] [CrossRef]
- Minato, M.; Yamamoto, K.; Tsuji, J. Osmium tetraoxide catalyzed vicinal hydroxylation of higher olefins by using hexacyanoferrate(III) ion as a cooxidant. J. Org. Chem. 1999, 55, 766–768. [Google Scholar] [CrossRef]
- Martin, V.S.; Woodard, S.S.; Katsuki, T.; Yamada, Y.; Ikeda, M.; Sharpless, K.B. Kinetic resolution of racemic allylic alcohols by enantioselective epoxidation. A route to substances of absolute enantiomeric purity? J. Am. Chem. Soc. 1981, 103, 6237–6240. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispino, G.A.; Hartung, J.; Jeong, K.S.; Kwong, H.L.; Morikawa, K.; Wang, Z.M. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768–2771. [Google Scholar] [CrossRef]
- Ogino, Y.; Chen, H.; Kwong, H.L.; Sharpless, K.B. On the timing of hydrolysis / reoxidation in the osmium-catalyzed asymmetric dihydroxylation of olefins using potassium ferricyanide as the reoxidant. Tetrahedron Lett. 1991, 32, 3965–3968. [Google Scholar] [CrossRef]
- Hofmann, K.A. Sauerstoff-übertragung durch Osmiumtetroxyd und Aktivierung von Chlorat-Lösungen. Ber. Dtsch. Chem. Ges. 1912, 45, 3329–3336. [Google Scholar] [CrossRef]
- Rheenen, V.V.; Kelly, R.C.; Cha, D.Y. An improved catalytic OsO4 oxidation of olefins to cis-1,2-glycols using tertiary amine oxides as the oxidant. Tetrahedron Lett. 1976, 17, 1973–1976. [Google Scholar] [CrossRef]
- Gao, Y. Osmium Tetroxide-t-Butyl Hydroperoxide. In Encyclopedia of Reagents for Organic Synthesis; John Wiley Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Mehltretter, G.M.; Döbler, C.; Sundermeier, U.; Beller, M. An improved version of the Sharpless asymmetric dihydroxylation. Tetrahedron Lett. 2000, 41, 8083–8087. [Google Scholar] [CrossRef]
- Hentges, S.G.; Sharpless, K.B. Asymmetric induction in the reaction of osmium tetroxide with olefins. J. Am. Chem. Soc. 1980, 102, 4263–4265. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Amberg, W.; Beller, M.; Chen, H.; Hartung, J.; Kawanami, Y.; Lubben, D.; Manoury, E.; Ogino, Y.; Shibata, T.; et al. New ligands double the scope of the catalytic asymmetric dihydroxylation of olefins. J. Org. Chem. 1991, 56, 4585–4588. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hajiabbasi, P. Recent advances in C-heteroatom bond forming by asymmetric Michael addition. Mol. Divers. 2014, 18, 411–439. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Asadi, S.; Lashkariani, B.M. Recent progress in asymmetric Biginelli reaction. Mol. Divers. 2013, 17, 389–407. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V. Recent advances in the application of the Oppolzer camphorsultam as a chiral auxiliary. Tetrahedron Asymmetry 2014, 25, 1061–1090. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E. Recent applications of the Suzuki reaction in total synthesis. Tetrahedron 2012, 68, 9145–9178. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E.; Azimian, F. Recent developments of the Stille reaction as a revolutionized method in total synthesis. Tetrahedron 2014, 70, 7–21. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E.; Nzari, N. Negishi coupling: An easy progress for C–C bond construction in total synthesis. Mol. Divers. 2014, 18, 441–472. [Google Scholar] [CrossRef]
- Heravi, M.M.; Hashemi, E.; Ghobadi, N. Development of Recent Total Syntheses Based on the Heck Reaction. Curr. Org. Syn. 2013, 17, 2192–2224. [Google Scholar] [CrossRef]
- Heravi, M.M.; Lashaki, T.B.; Poorahmad, N. Applications of Sharpless asymmetric epoxidation in total synthesis. Tetrahedron Asymmetry 2015, 26, 405–495. [Google Scholar] [CrossRef]
- Engler, E.M.; Andose, J.D.; Schleyer, V.R. Critical evaluation of molecular mechanics. J. Am. Chem. Soc. 1973, 95, 8005–8025. [Google Scholar] [CrossRef]
- Blagg, B.S.J.; Boger, D.L. Total synthesis of (+)-camptothecin. Tetrahedron 2002, 58, 6343–6349. [Google Scholar] [CrossRef]
- Reddy, J.S.; Rao, B.V. A Short, Efficient, and Stereoselective Total Synthesis of a Pyrrolidine Alkaloid: (−)-Codonopsinine. J. Org. Chem. 2007, 72, 2224–2227. [Google Scholar] [CrossRef] [PubMed]
- Ramulu, U.; Rajaram, S.; Ramesh, D.; Babu, K.S. Total synthesis of synargentolide B. Tetrahedron Asymmetry 2015, 26, 928–934. [Google Scholar] [CrossRef]
- Das, T.; Mahapatra, T.; Nanda, S. Total synthesis of stagonolide B. Tetrahedron Lett. 2012, 53, 1186–1189. [Google Scholar] [CrossRef]
- Ghosal, S.; Saini, K.S.; Razdan, S. Crinum alkaloids: Their chemistry and biology. Phytochemistry 1985, 24, 2141–2156. [Google Scholar] [CrossRef]
- Pettit, G.; Orr, B.; Ducki, S. Anticancer, pancratistatin, phosphate prodrugs, phosphorylation. Anti-Cancer Drug Design 2000, 15, 389–395. Available online: https://www.ingentaconnect.com/contentone/cog/antcan/2000/00000015/00000006/art00001 (accessed on 12 March 2022). [PubMed]
- Zhao, Y.; Zhu, Y.; Ma, G.; Wei, Q.; Yang, S.; Zeng, X.; Zhang, H.; Chen, J. Short, Enantioselective, gram-scale synthesis of (−)-zephyranthine. Chem. Sci. 2021, 12, 9452–9457. [Google Scholar] [CrossRef]
- Zhou, Z.F.; Menna, M.; Cai, Y.S.; Guo, Y.W. Polyacetylenes of Marine Origin: Chemistry and Bioactivity. Chem. Rev. 2015, 115, 1543–1596. [Google Scholar] [CrossRef] [PubMed]
- Hartung, J.; Kneuer, R. Synthesis of enantiopure (2R)-configured muscarine alkaloids via selective alkoxyl radical ring-closure reactions. Tetrahedron Asymmetry 2003, 14, 3019–3031. [Google Scholar] [CrossRef]
- Gehlawat, A.; Prakash, R.; Pandey, S.K. A Short and Efficient Enantioselective Synthesis of (+)-(2S,3S,5S)-epi-Muscarine. Chem. Sel. 2020, 5, 6373–6375. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.J.; Zhang, D.B.; Wen, J.; Zhao, X.W.; Li, Y.; Gao, K. An unusual indole alkaloid with anti-adenovirus and anti-HSV activities from Alstonia scholaris. Tetrahedron Lett. 2014, 55, 1815–1817. [Google Scholar] [CrossRef]
- Cai, Y.S.; Sarotti, A.M.; Zhou, T.L.; Huang, R.; Qiu, G.F.; Tian, C.K.; Miao, Z.H.; Mandi, A.; Kurtan, T.; Cao, S.G.; et al. Flabellipparicine, a flabelliformide-apparicine-type bisindol alkaloid from Tabernaemontana divaricate. J. Nat. Prod. 2018, 81, 1976–1983. [Google Scholar] [CrossRef]
- Pourroy, B.; Honore, S.; Pasquier, E.; Rey, B.V.; Kruczynski, A.; Briand, C.; Braguer, D. Antiangiogenic concentrations of vinflunine increase the interphase microtubule dynamics and decrease the motility of endothelial cells. Cancer Res. 2006, 66, 3256–3263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Song, M.; Ao, Y.L.; Li, Y.; Zou, X.Y.; Xu, J.; Wang, Y.; Zhang, D.M.; Zhang, X.Q.; Ye, W.C. Alstolarines A and B, Two Unusual Monoterpenoid Indole Alkaloids with Acetal Moiety from Alstonia scholaris. Org. Chem. Front. 2020, 7, 3468–3473. [Google Scholar] [CrossRef]
- Gossan, D.P.A.; Alabdul Magid, A.; Yao, P.A.K.; Behr, J.B.; Ahibo, A.C.; Djakouré, L.A.; Harakat, D.; Nazabadioko, L.V. Glycosidase inhibitors from the roots of Glyphaea brevis. Phytochemistry 2015, 109, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Shibano, M.; Tsukamoto, D.; Kusano, G. Polyhydroxylated Alkaloids with Lipophilic Moieties as Glycosidase Inhibitors from Higher Plants. Heterocycles 2002, 57, 1539–1553. Available online: https://cir.nii.ac.jp/crid/1521417755858432768 (accessed on 12 March 2022).
- Byatt, B.J.; Kato, A.; Pyne, S.G. Synthesis and Structural Revision of Glyphaeaside C. Org. Lett. 2021, 23, 4029–4033. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, R.; Covell, D.; Ransom, T.T.; Gustafson, K.R.; Beutler, J.A. Englerin A, a Selective Inhibitor of Renal Cancer Cell Growth, from Phyllanthus engleri. Org. Lett. 2009, 11, 57–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sourbier, C.; Scroggins, B.T.; Ratnayake, R.; Prince, T.L.; Lee, S.; Lee, M.J.; Nagy, P.L.; Lee, Y.H.; Trepel, J.B.; Beutler, J.A.; et al. Englerin A Stimulates PKCθ to Inhibit Insulin Signaling and to Simultaneously Activate HSF1: Pharmacologically Induced Synthetic Lethality. Cancer Cell 2013, 23, 228–237. [Google Scholar] [CrossRef] [Green Version]
- Mou, S.B.; Xiao, W.; Wang, H.Q.; Wang, S.J.; Xiang, Z. Syntheses of Epoxyguaiane Sesquiterpenes (−)-Englerin A, (−)-Oxyphyllol, (+)-Orientalol E, and (+)-Orientalol F: A Synthetic Biology Approach. Org. Lett. 2020, 22, 1976–1979. [Google Scholar] [CrossRef]
- Vichnewski, W.; Sarti, S.J.; Gilbert, B.; Herz, W. Goyazensolide, a Schistosomicidal Heliangolide from Eremanthus-Goyazensis. Phytochemistry 1976, 15, 191–193. [Google Scholar] [CrossRef]
- Acuna, U.M.; Shen, Q.; Ren, Y.; Lantvit, D.D.; Wittwer, J.A.; Kinghorn, A.D.; Swanson, S.M.; Carcach de Blanco, E.J. Goyazensolide Induces Apoptosis in Cancer Cells in vitro and in vivo. Int. J. Cancer Res. 2013, 9, 36–53. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4303185/ (accessed on 12 March 2022). [CrossRef] [PubMed] [Green Version]
- Liu, W.; Patouret, R.; Barleunga, S.; Plank, M.; Loewith, R.; Winssinger, N. Identification of a Covalent Importin-5 Inhibitor, Goyazensolide, from a Collective Synthesis of Furanoheliangolides. ACS Cent. Sci. 2021, 7, 954–962. [Google Scholar] [CrossRef]
- Maurer, B.; Grieder, A. Sesquiterpenoids from Costus Root Oil (Saussurea lappa CLARKE). Helvetica 1977, 60, 2177–2190. [Google Scholar] [CrossRef]
- Seiichi, T.; Takumichi, S.; Kiyohiro, S.; Masashi, A.; Kunio, O. Enantiodivergent Route to the Aromatic Bisabolane Sesquiterpenes via a Chiral Acetylene Alcohol. Chem. Lett. 1989, 18, 1781–1784. [Google Scholar] [CrossRef]
- Yajima, A.; Shirakawa, I.; Shiotani, N.; Ueda, K.; Murakawa, H.; Saito, T.; Katsuta, R.; Ishigami, K. Practical synthesis of aromatic bisabolanes: Synthesis of 1,3,5-bisabolatrien-7-ol, peniciaculin A and B, and hydroxysydonic acid. Tetrahedron 2021, 92, 132253. [Google Scholar] [CrossRef]
- Ushakov, D.B.; Navickas, V.; Strobele, M.; Mossmer, C.M.; Sasse, F.; Maier, M.E. Total Synthesis and Biological Evaluation of (−)-9-Deoxy-englerin A. Org. Lett. 2011, 13, 2090–2093. [Google Scholar] [CrossRef]
- Peng, G.P.; Tian, G.; Huang, X.F.; Lou, F.C. Guaiane-type sesquiterpenoids from Alisma orientalis. Phytochemistry 2003, 63, 877–881. [Google Scholar] [CrossRef]
- Palli, K.K.; Anugu, R.R.; Chandrasekhar, S. Total Synthesis of (−)-4- epi -Englerin A. Eur. J. Chem. 2021, 22, 31990–33196. [Google Scholar] [CrossRef]
- Alvarado, R.B.H.; Mazon, A.M.; Ortega, A.; Mayorga, K.M. DARK Classics in Chemical Neuroscience: Salvinorin A. ACS Chem. Neurosci. 2020, 11, 3979–3992. [Google Scholar] [CrossRef]
- Zimdars, P.; Wang, Y.; Metz, P. A Protecting-Group-Free Synthesis of (-)Salvinorin A. Chem. A Eur. J. 2021, 27, 7968–7973. [Google Scholar] [CrossRef]
- Xiao, W.L.; Li, R.T.; Huang, S.X.; Pu, J.X.; Sun, H.D. Triterpenoids from the Schisandraceae family. Nat. Prod. Rep. 2008, 25, 871–891. [Google Scholar] [CrossRef]
- Li, X.; Cheong, P.H.Y.; Carter, R.G. Schinortriterpenoids: A case study in synthetic design. Angew. Chem. Int. Ed. 2017, 56, 1704–1718. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, B.; He, X.; Gui, J. Bioinspired Synthesis of Nortriterpenoid Propindilactone G. J. Am. Chem. Soc. 2020, 142, 5007–5012. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.M.G.D.; Fo, S.R. Meroterpenes from Penicillium sp found in association with Melia azedarach. Phytochemistry 2002, 61, 907–912. [Google Scholar] [CrossRef]
- Guo, C.J.; Knox, B.P.; Chiang, Y.M.; Lo, H.C.; Sanchez, J.F.; Lee, K.H.; Okaley, B.R.; Bruno, K.S.; Wang, C.C.C. Molecular Genetic Characterization of a Cluster in A. terreus for Biosynthesis of the Meroterpenoid Terretonin. Org. Lett. 2012, 14, 5684–5687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ji, Y.; Franzoni, I.; Guo, C.; Jia, H.; Hong, B.; Li, H. Enantioselective Total Synthesis of Berkeleyone A and Preaustinoids. Angew. Chem. Int. Ed. 2021, 60, 14869–14874. [Google Scholar] [CrossRef]
- She, G.; Xu, C.; Liu, B.; Shi, R. Two new monoterpenes from Mentha haplocalyx Briq. Helv. Chim. Acta 2010, 93, 2495–2498. [Google Scholar] [CrossRef]
- Konishi, S.; Mori, N.; Takikawa, H. Synthesis guided structure revision of the monoterpene alcohol isolated from Mentha haplocalyx. Biosci. Biotechnol. Biochem. 2019, 83, 391–399. [Google Scholar] [CrossRef]
- Konishi, S.; Ogura, Y.; Takikawa, H.; Watanabe, H. Asymmetric synthesis of trans-p-menth-3-ene-1,2,8-triol, the monoterpene isolated from herbal plants. Biosci. Biotechnol. Biochem. 2020, 84, 37–42. [Google Scholar] [CrossRef]
- Yan, C.; Wan, R.; Shi, Y. Molecular Mechanisms of pre-mRNA Splicing through Structural Biology of the Spliceosome. Cold Spring Harb. Perspect. Biol. 2019, 14, a032409. Available online: https://cshperspectives.cshlp.org/content/11/1/a032409.short (accessed on 7 April 2022). [CrossRef] [Green Version]
- Sakai, T.; Sameshima, T.; Matsufuji, M.; Kawamura, N.; Dobashi, K.; Mizui, Y. Pladienolides, New Substances from Culture of Streptomyces Platensis Mer-11107. I. Taxonomy, Fermentation, Isolation and Screening. J. Antibiot. 2004, 57, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Rhoades, D.; Rheingold, A.L.; O’Malley, B.W.; Wang, J. Expedient Total Syntheses of Pladienolide-Derived Spliceosome Modulators. J. Am. Chem. Soc. 2021, 143, 4915–4920. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Kim, Y.; In, Y.; Ishida, T.; Kan, Y.; Fujita, T.; Iwashita, T.; Tabata, H.; Onaka, H.; Furumai, T. Alchivemycin A, a Bioactive Polycyclic Polyketide with an Unprecedented Skeleton from Streptomyces sp. Org. Lett. 2010, 12, 3402–3405. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Liao, D.; Tian, X.; Lei, X. Access to the 2H-Tetrahydro-4,6-dioxo-1,2-oxazine Ring System from Nitrone via a Tandem Nucleophilic Addition and Transesterification Reaction. Org. Lett. 2016, 18, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Yang, S.; Ma, K.; Wang, X.; Lei, X. Stereoselective synthesis of the C16–C25 fragment of alchivemycins A and B. Tetrahedron Lett. 2021, 74, 153156. [Google Scholar] [CrossRef]
- Trost, B.M.; Knopf, J.D.; Brindle, C.S. Synthetic Strategies Employed for the Construction of Fostriecin and Related Natural Products. Chem. Rev. 2016, 116, 15035–15088. [Google Scholar] [CrossRef] [Green Version]
- Just, G.; O’Connor, B. Synthesis of the 5R,8R,9S,11R Dephosphorylated Derivative of CI-920, a Novel Antitumor Agent. Tetrahedron Lett. 1988, 29, 753–756. [Google Scholar] [CrossRef]
- Dong, G.; Li, B.; O’Doherty, G. Total and Formal Syntheses of Fostriecin. Org. Chem. Front. 2020, 7, 3608–3615. [Google Scholar] [CrossRef]
- Seibert, S.F.; Krick, A.; Eguereva, E.; Kehraus, S.; Konig, G.M. Ascospiroketals A and B, Unprecedented Cycloethers from the Marine-Derived Fungus Ascochyta salicorniae. Org. Lett. 2007, 9, 239–242. [Google Scholar] [CrossRef]
- Hara, Y.; Kamaike, K.; Ota, K.; Miyaoka, H. Total Synthesis of Ascospiroketal B. Synlett 2020, 31, 1730–1734. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Baran, P.S. Total Synthesis of Eudesmane Terpenes by Site-Selective C−H Oxidations. Nature 2009, 459, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Wilde, N.C.; Isomura, M.; Mendoza, A.; Baran, P.S. Two-Phase Synthesis of (−)-Taxuyunnanine D. J. Am. Chem. Soc. 2014, 136, 4909–4912. [Google Scholar] [CrossRef] [PubMed]
- Gayraud, O.; Laroche, B.; Casaretto, N.; Nay, B. Synthesis of a Biomimetic Tetracyclic Precursor of Aspochalasins and Formal Synthesis of Trichoderone A. Org. Lett. 2021, 23, 5755–5760. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, K.; Minamida, M.; Akakabe, M.; Tsuda, M.; Konishi, Y.; Tominaga, A.; Tsuda, M.; Fukushi, E.; Kawabata, J. Amphirionin-2, a Novel Linear Polyketide with Potent Cytotoxic Activity from a Marine Dinoflagellate Amphidinium Species. Bioorg. Med. Chem. Lett. 2015, 25, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Mizukami, D.; Sugai, T.; Tsuda, M.; Fuwa, H. Total Synthesis and Complete Configurational Assignment of Amphirionin-2. Chem. Sci. 2021, 12, 872–879. [Google Scholar] [CrossRef]
- Saha, D.; Mandal, G.H.; Goswami, R.K. Asymmetric Total Synthesis of Amphirionin-2. J. Org. Chem. 2021, 86, 10006–10022. [Google Scholar] [CrossRef]
- Wilton, J.H.; Cheney, D.C.; Hokanson, G.C.; French, J.C.; Cunheng, H.; Clardy, J. A new dihydrobenz[a]anthraquinone antitumor antibiotic (PD 116,740). J. Org. Chem. 1985, 50, 3936–3938. [Google Scholar] [CrossRef]
- Rohr, J.; Thiericke, R. Angucycline Group Antibiotics. Nat. Prod. Rep. 1992, 9, 103–137. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Xie, T.; He, H.; Gao, S. Asymmetric Total Synthesis of PD-116740. Org. Lett. 2021, 23, 469–473. [Google Scholar] [CrossRef]
- Trost, B.M.; Wang, Y.; Buckl, A.K.; Huang, Z.; Nguyen, M.H.; Kuzmina, O. Total synthesis of bryostatin 3. Science 2020, 368, 1007–1011. [Google Scholar] [CrossRef]
- Chen, Q.-H.; Kingston, D.G.I. Zampanolide and dactylolide: Cytotoxic tubulin-assembly agents and promising anticancer leads. Nat. Prod. Rep. 2014, 31, 1202–1226. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Jiang, Z.; Zhang, Q.; Wang, G.; Chen, Q.H. New Zampanolide Mimics: Design, Synthesis, and Antiproliferative Evaluation. Molecules 2020, 25, 362. [Google Scholar] [CrossRef] [Green Version]
- Carreira, E.M.; Pfaff, P. Total Synthesis of (+)-Formosalides A and B. Synfacts 2020, 16, 1387. [Google Scholar] [CrossRef]
- Sekiguchi, J.; Kuroda, H.; Yamada, Y.; Okada, H. Structure of patulolide a, a new macrolide from penicillium urticae mutants. Tetrahedron Lett. 1985, 26, 2341–2342. [Google Scholar] [CrossRef]
- Rodpha, D.; Sekiguchi, J.; Yamada, Y. New macrolides from Penicillium Urticae mutant S11R59. J. Antibiot. 1986, 39, 629–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paratapareddy, B.; Sreenevasulu, R.; Venkata, M.; Rao, B.; Raju, R.R. Stereoselective total synthesis of Patulolide C. Nat. Prod. Res. 2020, 34, 2760–2764. [Google Scholar] [CrossRef]
- Khalil, M.W.; Sasse, F.; Lunsdorf, H.; Elnakady, Y.A.; Reichenbach, H. Mechanism of Action of Tubulysin, an Antimitotic Peptide from Myxobacteria. Chembiochem 2006, 7, 678–683. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Erande, R.D.; Yin, J.; Vourloumis, D.; Aujay, M.; Sandoval, J.; Munneke, S.; Gavrilyuk, J. Improved Total Synthesis of Tubulysins and Design, Synthesis, and Biological Evaluation of New Tubulysins with Highly Potent Cytotoxicities against Cancer Cells as Potential Payloads for Antibody-Drug Conjugates. J. Am. Chem. Soc. 2018, 140, 3690–3711. [Google Scholar] [CrossRef]
- Reddy, R.B.; Krishnan, M.A.; Chelvam, V. Synthesis of tubuvaline (Tuv) fragment of tubulysin via diastereoselective diydroxylation of homoallylamine. Syn. Commun. 2021, 51, 797–809. [Google Scholar] [CrossRef]
- Schulz, B.; Boyle, C.; Draeger, S.; Rommert, A.K.; Krohn, K. Endophytic fungi: A source of novel biologically active secondary metabolites. Mycol. Res. 2002, 106, 996–1004. [Google Scholar] [CrossRef]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of Natural Products on Developing New Anti-Cancer Agents. Chem. Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Paul, D.; Goswami, R.K. Cyclodepsipeptide Alveolaride C: Total Synthesis and Structural Assignment. Chem. Sci. 2020, 11, 11259–11265. [Google Scholar] [CrossRef] [PubMed]
- Govindachari, T.R.; Kumari, K.G.N.; Suresh, G. Triterpenoids from Walsura piscidia. Phytochemistry 1995, 39, 167–170. [Google Scholar] [CrossRef]
- Ramana, L.V.; Apparao, K.M.C.; Rao, B.N.; Rao, S.A. Isolation & First Total Asymmetric Synthesis of New Phenolic Constituent: Isolation From Walsura Trifoliata. Eur. J. Pharm. Med. Res. 2021, 8, 616–621. [Google Scholar]
- Rinaldo, D.; Batista, J.M., Jr.; Rodrigues, J.; Benfatti, A.C.; Rodrigues, C.M.; Santos, L.C.D.; Furlan, M.; Vilegas, W. Determination of catechin diastereomers from the leaves of Byrsonima species using chiral HPLC-PAD-CD. Chirality 2010, 22, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Chung, S.; Song, M.-Y.; Lim, S.; Shin, H.; Hur, J.; Kwon, H.; Suh, Y.-G.; Kim, E.-H.; Shin, D.; et al. Efficient and Divergent Enantioselective Syntheses of DHPVs and Anti-Inflammatory Effect on IEC-6 Cells. Molecules 2020, 25, 2215. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, C.V.; Cai, S.X.; Peng, J.N.; Robles, A.J.; Hartley, R.M.; Powell, D.R.; Du, L.; Cichewicz, R.H.; Mooberry, S.L. Texas native plants yield compounds with cytotoxic activities against prostate cancer cells. J. Nat. Prod. 2016, 79, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Zhu, Z.; Ding, Y.; Li, G.; Li, N.; Shen, T. Synthesis and Cytotoxic Evaluation of Sanjoseolide and Representative Analogues. ACS Omega 2020, 5, 33478–33483. [Google Scholar] [CrossRef]
- Hikino, H.; Taguchi, T.; Fujimura, H.; Hiramatsu, Y. Antiinflammatory Principles of Caesalpinia Sappan Wood and of Haematoxylon Campechianum Wood. Plant Med. 1977, 31, 214–220. [Google Scholar] [CrossRef]
- Huang, S.; Ou, W.; Li, W.; Xiao, H.; Pang, Y.; Zhou, Y.; Wang, X.; Yang, X.; Wang, L. A total synthesis of (+)-brazilin. Tetrahedron Lett. 2020, 61, 152052. [Google Scholar] [CrossRef]
- Jeong, J.Y.; Jo, Y.H.; Lee, K.Y.; Do, S.G.; Hwang, B.Y.; Lee, M.K. Optimization of pancreatic lipase inhibition by Cudrania tricuspidata fruits using response surface methodology. Bioorg. Med. Chem. Lett. 2014, 24, 2329–2333. [Google Scholar] [CrossRef]
- Lu, Q.; Harmalkar, D.S.; Quan, G.; Kwon, H.; Cho, J.; Choi, Y.; Lee, D.; Lee, K. Total Synthesis of the Neuroprotective Agent Cudraisoflavone J. J. Nat. Prod. 2021, 84, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Brase, S.; Encinas, A.; Keck, J.; Nising, C.F. Chemistry and Biology of Mycotoxins and Related Fungal Metabolites. Chem. Rev. 2009, 109, 3903–3990. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Zheng, C.; Chen, K.; He, H.; Gao, S. Asymmetric Total Synthesis of FD-594. Angew. Chem. Int. Ed. 2020, 132, 4390–4394. [Google Scholar] [CrossRef]
- Roman, G.N.; Jansen, N.B.; Hsiao, H.Y.; Tsao, G.T. Kinetic studies of the enzymatic isomerization of xylose. Enzym. Microb. Technol. 1985, 7, 129–133. [Google Scholar] [CrossRef]
- Kalagara, S.; Orozco, G.; Mito, S. The efficient synthesis of D-xylulose and formal synthesis of Syringolide 1. Tetrahedron Lett. 2020, 61, 152321. [Google Scholar] [CrossRef]
- Compain, P.; Martin, O.R. Iminosugars: From Synthesis to Therapeutic Applications; Wiley: Chichester, UK, 2007. [Google Scholar] [CrossRef]
- Stocker, B.L.; Dangerfield, E.; Win-Mason, A.L.; Haslett, G.W.; Timmer, S.M. Recent Developments in the Synthesis of Pyrrolidine-Containing Iminosugars. Eur. J. Org. Chem. 2010, 9, 1615–1637. [Google Scholar] [CrossRef]
- Angelis, M.D.; Primitivo, L.; Lucarini, C.; Agostinelli, S.; Sappino, C.; Ricelli, A.; Righi, G. Stereocontrolled total synthesis of iminosugar 1,4-dideoxy-1,4-imino-D-iditol. Carbohydr. Res. 2020, 492, 108028. [Google Scholar] [CrossRef]
- Tatsuta, K.; Hosokawa, S. Total Synthesis of Selected Bioactive Natural Products: Illustration of Strategy and Design. Chem. Rev. 2005, 105, 4707–4729. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.; Hosokawa, S. Stereoselective Total Synthesis of the Dimeric Naphthoquinonopyrano-γ-lactone (−)-Crisamicin A: Introducing the Dimerization Site by a Late-Stage Hartwig Borylation. Org. Lett. 2020, 22, 3607–3612. [Google Scholar] [CrossRef]
- Dong, L.; Gordon, V.A.; Grange, R.L.; Johns, J.; Parsons, P.G.; Porzelle, A.; Reddell, P.; Schill, H.; Williams, C.M. Structure and Absolute Stereochemistry of the Anticancer Agent EBC-23 from the Australian Rainforest. J. Am. Chem. Soc. 2008, 130, 15262–15263. [Google Scholar] [CrossRef]
- Dong, L.; Schill, H.; Grange, R.L.; Porzelle, A.; Johns, J.P.; Parsons, P.G.; Gordon, V.A.; Reddell, P.W.; Williams, C.M. Anticancer Agents from the Australian Tropical Rainforest: Spiroacetals EBC-23, 24, 25, 72, 73, 75 and 76. Chem. A Eur. J. 2009, 15, 11307–11318. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Hsu, C.S. Enantioselective Total Synthesis of (+)-EBC-23, a Potent Anticancer Agent from the Australian Rainforest. J. Org. Chem. 2021, 86, 6351–6360. [Google Scholar] [CrossRef]
- Rao, N.N.; Meshram, H.M. Protection-free, short, and stereoselective synthesis of ieodomycin A and B. Tetrahedron Lett. 2013, 54, 4544–4546. [Google Scholar] [CrossRef]
- Choi, D.B.; Choi, H.; Lee, J.; Lee, Y.J.; Lee, H.S.; Joo, J.M.; Lee, J.S. Eantioselective Total Synthesis of (+)-Ieodomycin A, (+)-Ieodomycin B, and Their Three Stereoisomers. Org. Bio. Chem. 2020, 18, 9227–9230. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.M.; Feng, Y.F.; Gao, H.; Zhang, X.; Tang, J.S.; Yao, X.S. Three new bis-styryllactones from Goniothalamus cheliensis. Fitoterapia 2014, 82, 524–527. [Google Scholar] [CrossRef]
- Kovacevic, I.; Kesic, J.; Popsavin, M.; Francuz, J.; Kojic, V.; Jakimov, D.; Rodic, M.V.; Zelenovic, B.S.; Benedekovic, G.; Popsavin, V. Asymmetric synthesis and biological evaluation of (+)-cardiobutanolide, (−)-3-deoxycardiobutanolide and analogues as antiproliferative agents. Tetrahedron 2021, 97, 132408. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Bethi, V. A Concise Synthesis of the Key Tetrahydrofuran Moieties of Caruifolin A and EBC-342. Eur. J. Org. Chem. 2020, 2020, 6922–6928. [Google Scholar] [CrossRef]
- Kobayashi, J. Search for New Bioactive Marine Natural Products and Application to Drug Development. Chem. Pharm. Bull. 2016, 64, 1079–1083. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, J.; Tsuda, M. Amphidinolides, bioactive macrolides from symbiotic marine dinoflagellates. Nat. Prod. Rep. 2004, 21, 77–93. [Google Scholar] [CrossRef]
- Gajula, S.; Reddy, A.V.V.; Reddy, D.P.; Yadav, J.S.; Mohapatra, D.K. Stereoselective Synthesis of the C1–C16 Fragment of the Purported Structure of Formosalide B. ACS Omega 2020, 5, 10217–10224. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Kovela, S.; Osswald, H.L.; Amano, M.; Aoki, M.; Agniswamy, J.; Wang, Y.F.; Weber, I.T.; Mitsuya, H. Synthesis of an HIV-1 Protease Inhibitor. J. Med. Chem. 2020, 63, 4867–4879. [Google Scholar] [CrossRef]
- Alali, F.Q.; Liu, X.-X.; McLaughlin, J.L. Annonaceous Aceotgenins: Recent Progress. J. Nat. Prod. 1999, 62, 504–540. [Google Scholar] [CrossRef]
- Shi, G.; Kozlowski, J.F.; Schwedler, J.T.; Wood, K.V.; MacDougal, J.M.; MacLaughlin, J.L. Muconin and Mucoxin: Additional Nonclassical Bioactive Acetogenins from Rollinia mucosa. J. Org. Chem. 1996, 61, 7988–7989. [Google Scholar] [CrossRef]
- Sugimoto, M.; Nosho, S.; Hikosaka, G.; Hattori, Y.; Umezawa, K.; Kawamura, A.; Makabe, H. Synthesis of (+)-Muconin via Diastereoselective Oxypalladation. J. Org. Chem. 2021, 86, 4859–4866. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Seletsky, B.M.; Palme, M.H.; Lydon, P.J.; Singer, L.A.; Chage, C.E.; Lemelin, C.A.; Shen, Y.; Davis, H.; Tremblay, L.; et al. Macrocyclic ketone analogues of halichondrin B. Bioorg. Med. Chem. Lett. 2004, 14, 5551–5554. [Google Scholar] [CrossRef]
- Senapati, S.; Ramana, C.V. A Concise/catalytic approach for the construction of the C14–C28 fragment of eribulin. Org. Biomol. Chem. 2021, 19, 4542–4550. [Google Scholar] [CrossRef]
- Shimizu, Y. Microalgal metabolites. Chem. Rev. 1993, 93, 1685–1698. [Google Scholar] [CrossRef]
- Oishi, T. Structure Determination, Chemical Synthesis, and Evaluation of Biological Activity of Super Carbon Chain Natural Products. Bull. Chem. Soc. Jpn. 2020, 93, 1350–1360. [Google Scholar] [CrossRef]
- Becker, H.; Soler, M.A.; Sharpless, K.B. Selective Asymmetric Dihydroxylation of Polyenes. Tetrahedron 1995, 51, 1345–1376. [Google Scholar] [CrossRef]
- Luo, J.; Xiang, J.Y.; Yuan, H.Y.; Wu, J.Q.; Li, H.Z.; Shen, Y.H.; Xu, M. Isolation, synthesis and absolute configuration of 4,5-dihydroxypiperines improving behavioral disorder in AlCl3-induced dementia. Bioorg. Med. Chem. Lett. 2021, 42, 128057. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Numata, A.; Yamada, T.; Minoura, K.; Enomoto, S.; Konishi, K.; Nakai, M.; Matsuda, C.; Nomoto, K. Penostatins, novel cytotoxic metabolites from a Penicillium species separated from a green alga. Tetrahedron Lett. 1996, 37, 655–658. [Google Scholar] [CrossRef]
- Wang, J.; Cadena, M.A.M.; Tong, R. Asymmetric Total Syntheses of (+)-Penostatins A and C. Org. Lett. 2020, 22, 5074–5078. [Google Scholar] [CrossRef]
- Liu, M.; Sibi, F. Recent advances in the stereoselective synthesis of β-amino acids. Tetrahedron 2002, 58, 7991–8035. [Google Scholar] [CrossRef]
- Matsushima, Y.; Orita, M. Concise synthesis of the Taxol side chain and demethoxy-4-epicytoxazone via oxazoline formation through intramolecular benzylic substitution of a bis-trichloroacetimidate. Tetrahedron Lett. 2021, 73, 153095. [Google Scholar] [CrossRef]
- McKinney, J.D. In vivo veritas: The search for TB drug targets goes live. Nat. Med. 2000, 6, 1330–1333. Available online: https://www.nature.com/articles/nm1200_1330 (accessed on 7 April 2022). [CrossRef] [PubMed]
- Tsutsumi, T.; Matsumoto, M.; Iwasaki, H.; Tomisawa, K.; Komine, K.; Fukuda, H.; Eustache, J.; Jansen, R.; Hatakeyama, S.; Ishihara, J. Total Synthesis of Thuggacin cmc-A and Its Structure Determination. Org. Lett. 2021, 23, 5208–5212. [Google Scholar] [CrossRef]





















































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mushtaq, A.; Zahoor, A.F.; Bilal, M.; Hussain, S.M.; Irfan, M.; Akhtar, R.; Irfan, A.; Kotwica-Mojzych, K.; Mojzych, M. Sharpless Asymmetric Dihydroxylation: An Impressive Gadget for the Synthesis of Natural Products: A Review. Molecules 2023, 28, 2722. https://doi.org/10.3390/molecules28062722
Mushtaq A, Zahoor AF, Bilal M, Hussain SM, Irfan M, Akhtar R, Irfan A, Kotwica-Mojzych K, Mojzych M. Sharpless Asymmetric Dihydroxylation: An Impressive Gadget for the Synthesis of Natural Products: A Review. Molecules. 2023; 28(6):2722. https://doi.org/10.3390/molecules28062722
Chicago/Turabian StyleMushtaq, Aqsa, Ameer Fawad Zahoor, Muhammad Bilal, Syed Makhdoom Hussain, Muhammad Irfan, Rabia Akhtar, Ali Irfan, Katarzyna Kotwica-Mojzych, and Mariusz Mojzych. 2023. "Sharpless Asymmetric Dihydroxylation: An Impressive Gadget for the Synthesis of Natural Products: A Review" Molecules 28, no. 6: 2722. https://doi.org/10.3390/molecules28062722