
Revealing Medicinal Constituents of Bistorta vivipara Based on Non-Targeted Metabolomics and 16S rDNA Gene Sequencing Technology
 , ,
, ,
Abstract
:
1. Introduction
2. Results
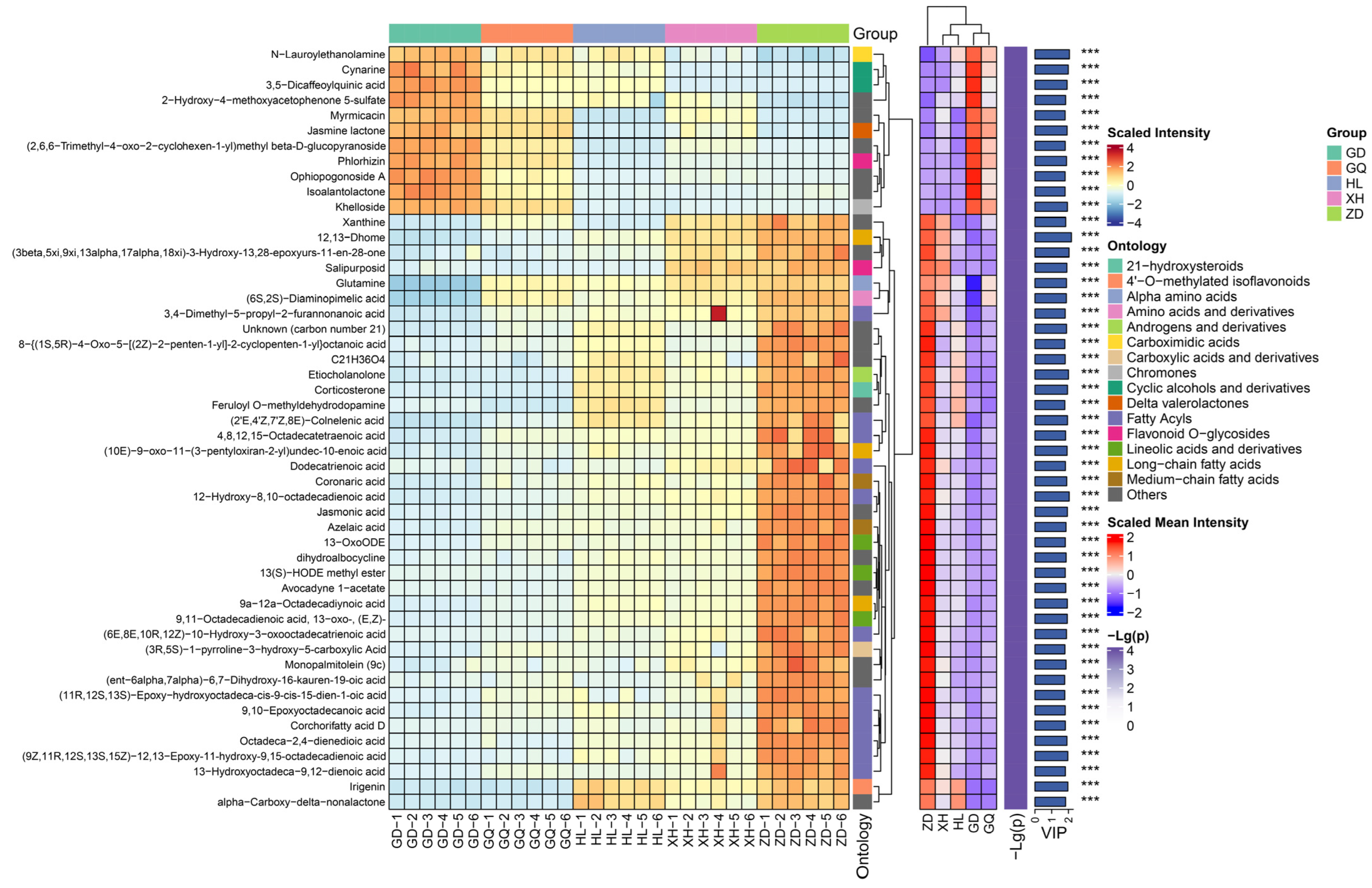
2.1. Metabolomics Analysis of the Roots of Bistorta vivipara
2.2. Identification of Differential Metabolites in the Roots of Bistorta vivipara from Different Sampling Areas
2.3. Differential Metabolite Functional Pathway Analysis
2.4. Analysis of Endophytic Bacterial Diversity in Bistorta vivipara Roots
2.5. Compositional Analysis of Endophytic Bacterial Communities in the Roots of Bistorta vivipara
2.6. Predictive Analysis of Microbial Function
2.7. Correlation Analysis of the Microbiomics and Metabolomics of Bistorta vivipara Root
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Untargeted Metabolomics Analysis
4.3. 16S Sequence Analysis
4.4. Correlation Analysis of Metabolomics and Microbiomics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mühlmann, O.; Bacher, M.; Peintner, U. Polygonum viviparum mycobionts on an alpine primary successional glacier forefront. Mycorrhiza 2008, 18, 87–95. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, H.; Zhang, F.; Chen, L.; Sun, W. Altitudinal phenotypic plasticity of leaf characteristics of Polygonum viviparum. Chin. J. Appl. Ecol. 2021, 32, 2070–2078. [Google Scholar]
- Zhen, D.; He, Z.; Qian, Z.; Xie, J.; Xu, M. Chemical Constituents from Stems and Leaves of Polygonum viviparum. J. Chin. Med. Mater. 2020, 43, 2172–2175. [Google Scholar]
- Bauert, R.M. Vivipary in Polygonum viviparum: An adaptation to cold climate? Nord. J. Bot. 1993, 13, 473–480. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, C.; Ren, Y.; Liu, S.; Wu, F. Response of Biological Characteristics and Medicinal Ouality of Wild Polygonum viviparum Community to Altitude and Aspect. Chin. Pharm. J. 2022, 57, 181–191. [Google Scholar]
- Qian, Z.; Cheng, X.; Wang, Q.; Huang, Q.; Jin, L.; Ma, Y.; Xie, J.; Li, D. On-line pre-column FRAP-based antioxidant reaction coupled with HPLC-DAD-TOF/MS for rapid screening of natural antioxidants from different parts of Polygonum viviparum. RSC Adv. 2023, 13, 9585–9594. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chen, J.; Wang, T.; Gao, C.; Li, Z.; Guo, L.; Xu, J.; Cheng, Y. Linking plant secondary metabolites and plant microbiomes: A review. Front. Plant Sci. 2021, 12, 621276. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Liu, D.; Chen, H.; Tang, D.; Zhao, Y. Metabolomics highlights pharmacological bioactivity and biochemical mechanism of traditional Chinese medicine. Chem. Biol. Interact. 2017, 273, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Arbona, V.; Manzi, M.; Ollas, C.; Gómez-Cadenas, A. Metabolomics as a tool to investigate abiotic stress tolerance in plants. Int. J. Mol. Sci. 2013, 14, 4885–4911. [Google Scholar] [CrossRef] [PubMed]
- Dahmani, I.; Qin, K.; Zhang, Y.; Ferni, A. The formation and function of plant metabolons. Plant J. 2023, 114, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wen, K.; Ruan, X.; Zhao, Y.; Wei, F.; Wang, Q. Response of plant secondary metabolites to environmental factors. Molecules 2018, 23, 762. [Google Scholar] [CrossRef]
- Li, Z.; Xiong, K.; Wen, W.; Li, L.; Xu, D. Functional endophytes regulating plant secondary metabolism: Current status, prospects and applications. Int. J. Mol. Sci. 2023, 24, 1153. [Google Scholar] [CrossRef] [PubMed]
- Amini, H.; Naghavi, M.; Shen, T.; Wang, Y.; Nasiri, J.; Khan, I.; Fiehn, O.; Zerbe, P.; Maloof, J. Tissue-specific transcriptome analysis reveals candidate genes for terpenoid and phenylpropanoid metabolism in the medicinal plant Ferula assafoetida. G3 Genes Genomes Genet. 2019, 9, 807–816. [Google Scholar] [CrossRef] [PubMed]
- JEE, H.; CHANG, K.; PARK, S.; KIM, K.; PAIK, A. Morphological characterization, chemical components, and biofunctional activities of Panax ginseng, Panax quinquefolium, and Panax notoginseng roots: A comparative study. Food Rev. Int. 2014, 30, 91–111. [Google Scholar] [CrossRef]
- Wei, G.; Chen, Z.; Wang, B.; Wei, F.; Zhang, G.; Wan, Y.; Zhu, G.; Zhou, Y.; Zhao, Q.; He, M.; et al. Endophytes isolated from Panax notoginseng converted ginsenosides. Microb. Biotechnol. 2021, 14, 1730–1746. [Google Scholar] [CrossRef]
- Li, L.; Su, C.; Chen, X.; Wang, Q.; Jiao, W.; Luo, H.; Tang, J.; Wang, W.; Li, S.; Guo, S. Chlorogenic acids in cardiovascular disease: A review of dietary consumption, pharmacology, and pharmacokinetics. J. Agric. Food Chem. 2020, 68, 6464–6484. [Google Scholar] [CrossRef]
- Xue, H.; Wei, M.; Ji, L. Chlorogenic Acids: A pharmacological systematic review on their hepatoprotective effects. Phytomedicine 2023, 118, 154961. [Google Scholar] [CrossRef]
- Roodi, D.; Millner, J.; McGill, C.; Johnson, R.; Jauregui, R.; Card, S. Methylobacterium, a major component of the culturable bacterial endophyte community of wild Brassica seed. PeerJ 2020, 8, e9514. [Google Scholar] [CrossRef]
- Li, R.; Duan, W.; Ran, Z.; Chen, X.; Yu, H.; Fang, L.; Guo, L.; Zhou, J. Diversity and correlation analysis of endophytes and metabolites of Panax quinquefolius L. in various tissues. BMC Plant Biol. 2023, 23, 275. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, M.; Zu, M.; Zuo, J.; Yuan, Y. Comprehensive Microbiome and Metabolome Analyses Reveal the Medicinal Components of Paeonia lactiflora. Plants 2023, 12, 1612. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Chen, L.; Ma, J.; Mo, J.; He, L.; Liang, Q.; Peng, G.; Tan, Z. Biological functions of endophytic bacteria in Robinia pseudoacacia ‘Hongsen’. Front. Microbiol. 2023, 14, 1128727. [Google Scholar] [CrossRef]
- Frank, A.; Guzmán, J.; Shay, J. Transmission of bacterial endophytes. Microorganisms 2017, 5, 70. [Google Scholar] [CrossRef]
- Wen, C.; Wu, Y.; Tian, X. Recent advances and issues on the endophyte. Chin. J. Ecol. 2004, 2, 86–91. [Google Scholar]
- Henning, J.; Weston, D.; Pelletier, D.; Timm, C.; Jawdy, S.; Classen, T.A. Root bacterial endophytes alter plant phenotype, but not physiology. PeerJ 2016, 4, e2606. [Google Scholar] [CrossRef]
- Fu, S.; Deng, Y.; Zou, K.; Zhang, S.; Liu, X.; Liang, Y. Flavonoids affect the endophytic bacterial community in Ginkgo biloba leaves with increasing altitude. Front. Plant Sci. 2022, 13, 982771. [Google Scholar] [CrossRef]
- Friesen, M.; Porter, S.; Stark, S.; Wettberg, E.; Sachs, J.; Martinez-Romero, E. Microbially mediated plant functional traits. Annu. Rev. Ecol. Evol. Syst. 2011, 42, 23–46. [Google Scholar] [CrossRef]
- Luo, X. Study on the Diversity of Endophyte from Roots of Vaccinium dunalianum and the Chemical Biology of the Dominant Fungus. Ph.D. Thesis, Southwest Forestry University, Kunming, China, 2022. [Google Scholar]
- Chi, Y.; Ma, X.; Wu, J.; Wang, R.; Zhang, X.; Chu, S.; Zhang, D.; Zhou, P. Plant growth promoting endophyte promotes cadmium accumulation in Solanum nigrum L. by regulating plant homeostasis. J. Hazard. Mater. 2023, 457, 131866. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Agarwal, S.; Verma, K.; Bhardwaj, R.; Mathur, V. Therapeutic compounds from medicinal plant endophytes: Molecular and metabolic adaptations. J. Appl. Microbiol. 2023, 134, lxad074. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Choi, J.; Baek, K. Control of Foodborne Pathogenic Bacteria by Endophytic Bacteria Isolated from Ginkgo biloba L. Foodborne Pathog. Dis. 2019, 16, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Jain, R.; Bhardwaj, P.; Thakur, A.; Kumari, M.; Bhushan, S.; Kumar, S. Plant Probiotics–Endophytes pivotal to plant health. Microbiol. Res. 2022, 263, 127148. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Zhang, C.; Ju, X.; Xiong, Y.; Xing, K.; Qin, S. Community composition and metabolic potential of endophytic actinobacteria from coastal salt marsh plants in Jiangsu, China. Front. Microbiol. 2019, 10, 1063. [Google Scholar] [CrossRef]
- Strobel, G.; Daisy, B. Bioprospecting for microbial endophytes and their natural products. Microbiol. Mol. Biol. Rev. 2003, 67, 491–502. [Google Scholar] [CrossRef]
- Gond, S.; Bergen, M.; Torres, M.; White, J.F., Jr. Endophytic Bacillus spp. produce antifungal lipopeptides and induce host defence gene expression in maize. Microbiol. Res. 2015, 172, 79–87. [Google Scholar] [CrossRef]
- Chen, J.; Feng, W.; Yan, H.; Liu, P.; Zhou, G.; Guo, S.; Yu, G.; Duan, J. Explore the interaction between root metabolism and rhizosphere microbiota during the growth of Angelica sinensis. Front. Plant Sci. 2022, 13, 1005711. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, H.; Chen, Z.; Deng, C.; Zhou, L.; Chen, S.; Kang, J.; Chen, Y.; He, S.; Zhou, Z. Identification of a novel plasma metabolite panel as diagnostic biomarker for hepatocellular carcinoma. Clin. Chim. Acta 2023, 543, 117302. [Google Scholar] [CrossRef]
- Kanne, C.; Reddy, V.; Sheehan, V. Rheological Effects of L-Glutamine in Patients with Sickle Cell Disease. Blood 2019, 134, 3567. [Google Scholar] [CrossRef]
- Wada, Y.; Totsune, E.; Mikami-Saito, Y.; Kikuchi, A.; Miyata, T.; Kure, S. A method for phenylalanine self-monitoring using phenylalanine ammonia-lyase and a pre-existing portable ammonia detection system. Mol. Genet. Metab. Rep. 2023, 35, 100970. [Google Scholar] [CrossRef]
- Liga, S.; Paul, C.; Péter, F. Flavonoids: Overview of Biosynthesis, Biological Activity, and Current Extraction Techniques. Plants 2023, 12, 2732. [Google Scholar] [CrossRef] [PubMed]
- Erlejman, A.; Verstraeten, S.; Fraga, C.; Oteiza, P. The interaction of flavonoids with membranes: Potential determinant of flavonoid antioxidant effects. Free Radic. Res. 2004, 38, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Sugimoto, Y.; Masuda, H.; Kamei, C. Antiallergic effect of flavonoid glycosides obtained from Mentha piperita L. Biol. Pharm. Bull. 2002, 25, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Q.; Ali, A.; Mukhtar, S.; Alsharif, M.; Parveen, H.; Sabere, A.; Nawi, M.; Khatib, A.; Siddiqui, M.; Umar, A. Medicinal potential of isoflavonoids: Polyphenols that may cure diabetes. Molecules 2020, 25, 5491. [Google Scholar] [CrossRef]
- Stanisławska, I.; Figat, R.; Kiss, A.; Bobrowska-Korczak, B. Essential Elements and Isoflavonoids in the Prevention of Prostate Cancer. Nutrients 2022, 14, 1225. [Google Scholar] [CrossRef]
- Cui, M.; Alhamshari, Y.; Cantwell, L.; EI-Haou, S.; Eptaminitaki, G.; Chang, M.; Abou-Assali, O.; Tan, H.; Xu, K.; Masotti, M.; et al. A benzopyran with antiarrhythmic activity is an inhibitor of Kir3. 1-containing potassium channels. J. Biol. Chem. 2021, 296, 100535. [Google Scholar] [CrossRef]
- Qian, Z.; Chen, L.; Wu, M.; Li, D. Rapid screening and characterization of natural antioxidants in Polygonum viviparum by an on-line system integrating the pressurised liquid micro-extraction, HPLC-DAD-QTOF-MS/MS analysis and antioxidant assay. J. Chromatogr. B 2020, 1137, 121926. [Google Scholar] [CrossRef]
- Brosnan, M.; Brosnan, J. Histidine Metabolism and Function. J. Nutr. 2020, 150, 2570S–2575S. [Google Scholar] [CrossRef]
- Li, P.; Ge, J.; Li, H. Lysine acetyltransferases and lysine deacetylases as targets for cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 96–115. [Google Scholar] [CrossRef]
- Choi, K. Nitrogen-Neutral Amino Acids Refinery: Deamination of Amino Acids for Bio-Alcohol and Ammonia Production. ChemBioEng Rev. 2021, 8, 213–226. [Google Scholar] [CrossRef]
- Chandel, N. Amino Acid Metabolism. Cold Spring Harb. Perspect. Biol. 2021, 13, a040584. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Yoshida, N.; Tsokos, G. Amino Acid Metabolism in Lupus. Front. Immunol. 2021, 12, 623844. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of Amino Acids in Cancer#13. Front. Cell Dev. Biol. 2021, 8, 603837. [Google Scholar] [PubMed]
- Chen, S. Characteristics of Root Zone Microbial Diversity and Isolation and Antibacterial Activity of Culturable Endophytes of Two Plants of Meconopsis Species; Tibet University: Lhasa, China, 2023. [Google Scholar]
- Chen, D.; Liu, H.; Yang, J.; Piao, X. Research Process on Active Constituents of Endophyte. Lishizhen Med. Mater. Medica Res. 2013, 24, 1730–1732. [Google Scholar]
- Wang, M.; Yang, S.; Liu, X.; Chen, Q. Root Characteristics of Plants Under Salt Stress and Their Interactions with Microorganisms. World For. Res. 2023, 36, 22–27. [Google Scholar]
- Wang, H.; Tang, Q.; Gu, M.; Zhang, Z.; Zhu, J. Composition and distribution pattern of endophytic community of Kalidium foliatum in Xinjiang’s saline-alkali regions. Agric. Res. Arid Areas 2023, 41, 257–266. [Google Scholar]
- Song, Z.; Ding, L.; Ma, B.; Li, W. Studies on the Population and Dynamic Analysis of Peanut Endophytes. J. Plant Prot. 1999, 26, 309–314. [Google Scholar]
- Zhang, Y.; Zhang, C.; Wang, Z.; Chen, Y.; Gang, C.; An, R.; Li, J. Vegetation dynamics and its driving forces from climate change and human activities in the Three-River Source Region, China from 1982 to 2012. Sci. Total Environ. 2016, 563–564, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Afzal, I.; Shinwari, Z.; Sikandar, S.; Shahzad, S. Plant beneficial endophytic bacteria: Mechanisms, diversity, host range and genetic determinants. Microbiol. Res. 2019, 221, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Mao, C.; Wan, Z.; Zhang, S.; Lu, H.; Mao, W.; Zhao, X.; Leng, L.; Bian, F. Effect of Storage Temperature on the Diversity ofEndophytic Bacteria in Phyllostachys nuda Shoots. J. Bamboo Res. 2023, 42, 48–55. [Google Scholar]
- Papik, J.; Folkmanova, M.; Polivkova-Majorova, M.; Suman, J.; Uhlik, O. The invisible life inside plants: Deciphering the riddles of endophytic bacterial diversity. Biotechnol. Adv. 2020, 44, 107614. [Google Scholar] [CrossRef]
- Naylor, D.; DeGraaf, S.; Purdom, E.; Coleman-Derr, D. Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 2017, 11, 2691–2704. [Google Scholar] [CrossRef]
- Gonzalo, G.; Colpa, D.; Habib, M.; Fraaije, M. Bacterial enzymes involved in lignin degradation. J. Biotechnol. 2016, 236, 110–119. [Google Scholar] [CrossRef]
- Miao, S.; Xia, Y.; Cui, J.; Wang, J.; Wang, M. Correlation analysis between differential metabolites and bacterial endophytes of Ephedra sinica in different years. Ind. Crops Prod. 2022, 175, 114250. [Google Scholar] [CrossRef]
- Song, X.; Zhang, J.; Li, D.; Peng, C. Nitrogen-fixing cyanobacteria have the potential to improve nitrogen use efficiency through the reduction of ammonia volatilization in red soil paddy fields. Soil Tillage Res. 2022, 217, 105274. [Google Scholar] [CrossRef]
- Chen, D.; Jia, L.; Hou, Q.; Zhao, X.; Sun, K. Analysis of Endophyte Diversity of Rheum palmatum from Different Production Areas in Gansu Province of China and the Association with Secondary Metabolite. Microorganisms 2021, 9, 978. [Google Scholar] [CrossRef]
- Langille, M.; Zaneveld, J.; Caporaso, J.; McDonald, D.; Knights, D.; Reyes, J.; Clemente, J.; Burkepile, D.; Thurber, R.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Song, X.; Wu, H.; Yin, Z.; Lian, M.; Yin, C. Endophytic bacteria isolated from Panax ginseng improves ginsenoside accumulation in adventitious ginseng root culture. Molecules 2017, 22, 837. [Google Scholar] [CrossRef]
- Jarocka-Karpowicz, I.; Markowska, A. Therapeutic Potential of Jasmonic Acid and Its Derivatives. Int. J. Mol. Sci. 2021, 22, 8437. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shi, X.; Tian, Y.; Zhai, S.; Liu, Y.; Xiong, Z.; Chu, S. An insight into novel therapeutic potentials of taxifolin. Front. Pharmacol. 2023, 14, 1173855. [Google Scholar] [CrossRef]
- Cai, C.; Liu, C.; Zhao, L.; Liu, H.; Li, W.; Guan, H.; Zhao, L.; Xiao, J. Effects of Taxifolin on Osteoclastogenesis in vitro and in vivo. Front. Pharmacol. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Xu, J.; Sun, S.; Zhang, W.; Dong, J.; Huang, C.; Wang, X.; Jia, M.; Yang, H.; Wang, Y.; Jiang, Y.; et al. Irigenin inhibits glioblastoma progression through suppressing YAP/β-catenin signaling. Front. Pharmacol. 2022, 13, 1027577. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Chao1 | Simpson | Shannon | Good’s Coverage |
|---|---|---|---|---|
| GD_1 | 322.661 | 0.488432 | 1.48113 | 0.999039 |
| GD_2 | 782.503 | 0.499047 | 2.43928 | 0.997733 |
| GD_3 | 411.038 | 0.400322 | 1.49202 | 0.999205 |
| GQ_1 | 218.223 | 0.406927 | 1.17572 | 0.999249 |
| GQ_2 | 307.265 | 0.452603 | 1.44211 | 0.999244 |
| GQ_3 | 157.634 | 0.493170 | 1.14908 | 0.999448 |
| HL_1 | 1803.710 | 0.820674 | 5.42361 | 0.995588 |
| HL_2 | 3341.240 | 0.969945 | 8.77754 | 0.994663 |
| HL_3 | 2368.240 | 0.895206 | 6.71797 | 0.994931 |
| XH_1 | 967.941 | 0.623918 | 3.17114 | 0.999507 |
| XH_2 | 1361.050 | 0.609018 | 3.55784 | 0.998728 |
| XH_3 | 1339.260 | 0.655782 | 3.81046 | 0.997838 |
| ZD_1 | 1751.590 | 0.901730 | 6.51395 | 0.996411 |
| ZD_2 | 1746.330 | 0.966805 | 7.55924 | 0.998378 |
| ZD_3 | 2269.150 | 0.990300 | 8.67914 | 0.997106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, H.; Tang, C.; Cao, Z.; Wang, T.; He, M.; Xiao, M.; Xiao, L.; Li, Y.; Li, X. Revealing Medicinal Constituents of Bistorta vivipara Based on Non-Targeted Metabolomics and 16S rDNA Gene Sequencing Technology. Molecules 2024, 29, 860. https://doi.org/10.3390/molecules29040860
He H, Tang C, Cao Z, Wang T, He M, Xiao M, Xiao L, Li Y, Li X. Revealing Medicinal Constituents of Bistorta vivipara Based on Non-Targeted Metabolomics and 16S rDNA Gene Sequencing Technology. Molecules. 2024; 29(4):860. https://doi.org/10.3390/molecules29040860
Chicago/Turabian StyleHe, Hui, Chuyu Tang, Zhengfei Cao, Tao Wang, Min He, Mengjun Xiao, Licheng Xiao, Yuling Li, and Xiuzhang Li. 2024. "Revealing Medicinal Constituents of Bistorta vivipara Based on Non-Targeted Metabolomics and 16S rDNA Gene Sequencing Technology" Molecules 29, no. 4: 860. https://doi.org/10.3390/molecules29040860