Complete Chloroplast Genomes and Comparative Analysis of Sequences Evolution among Seven Aristolochia (Aristolochiaceae) Medicinal Species


Abstract
:1. Introduction
2. Results
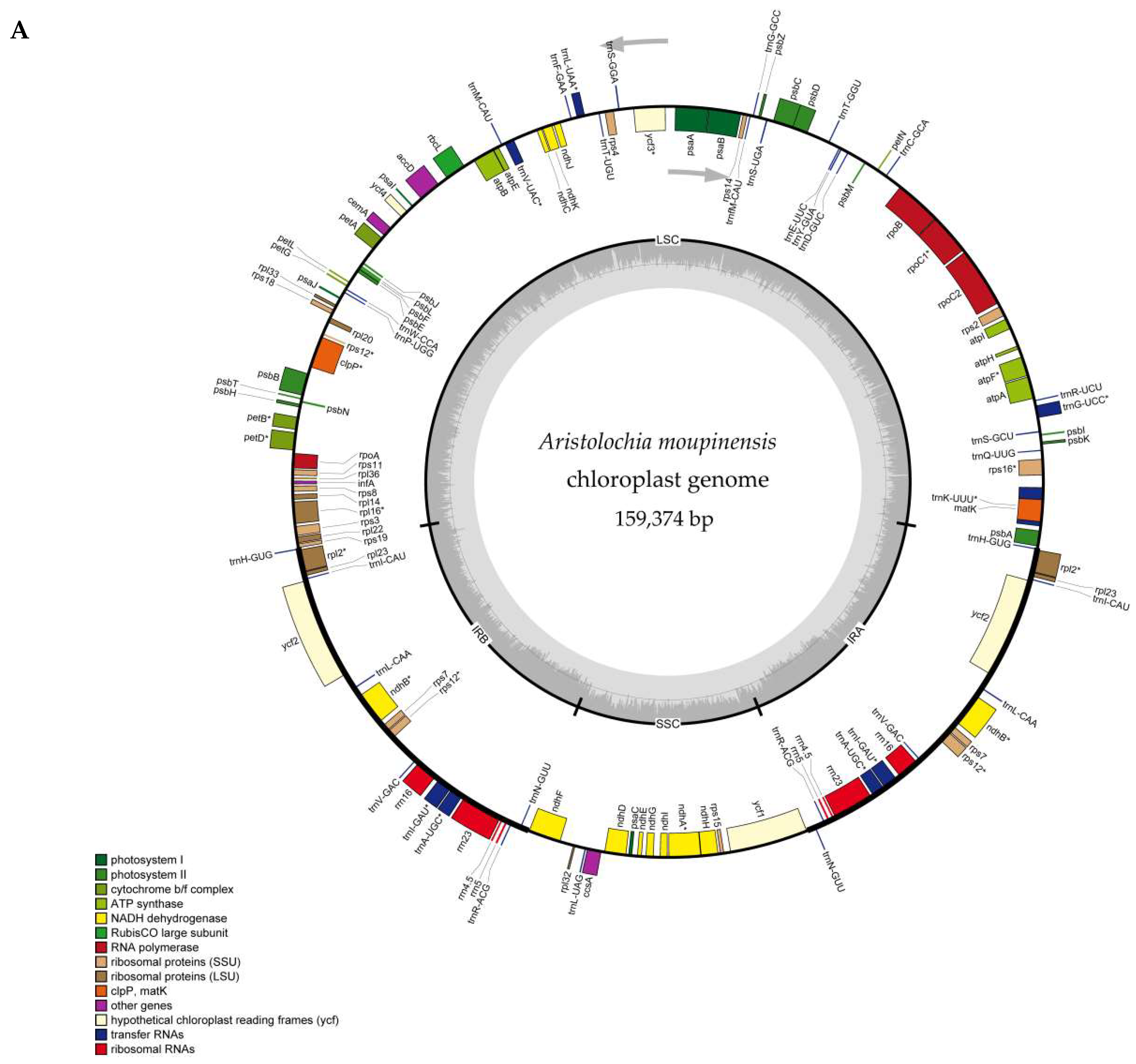
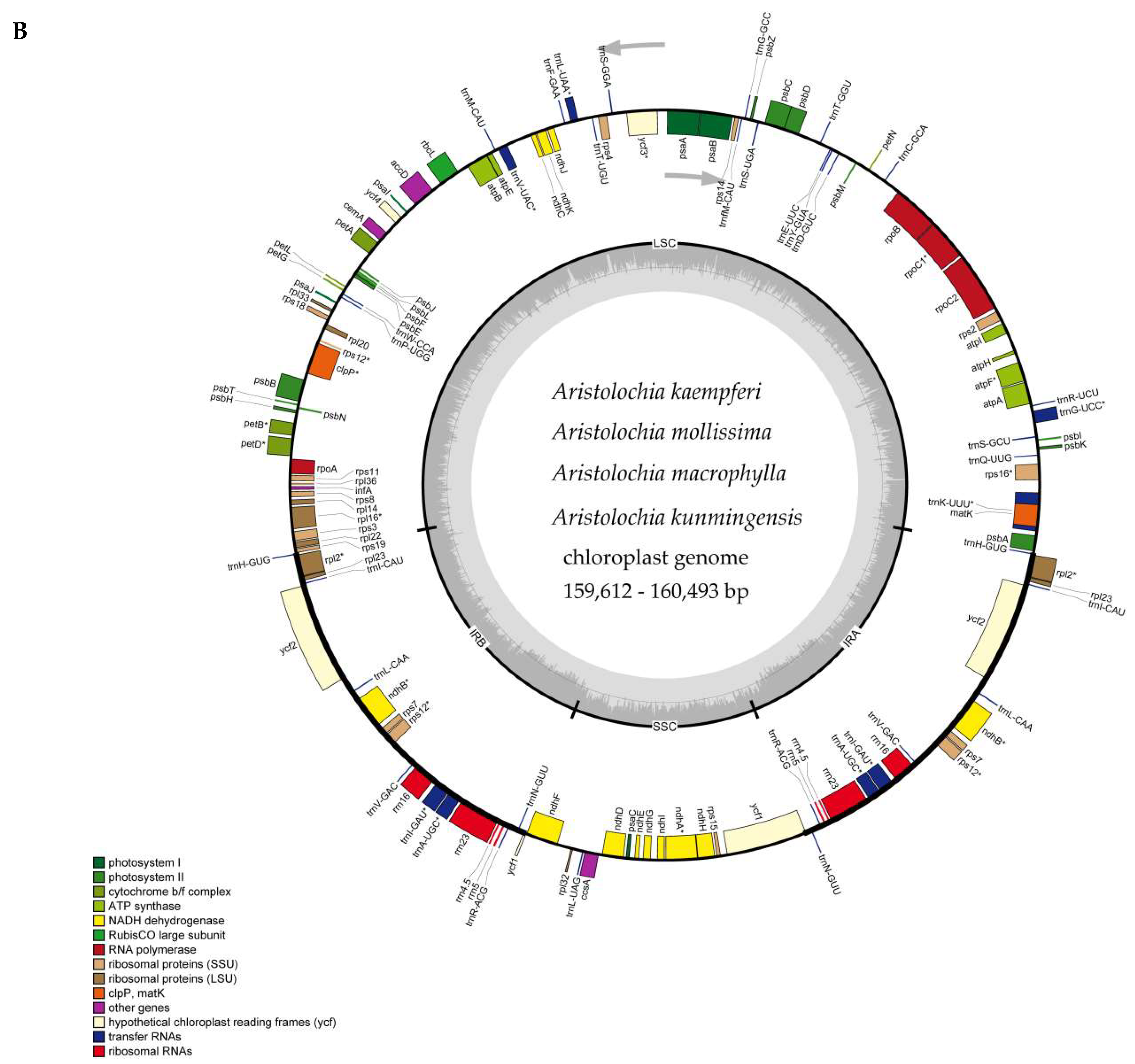
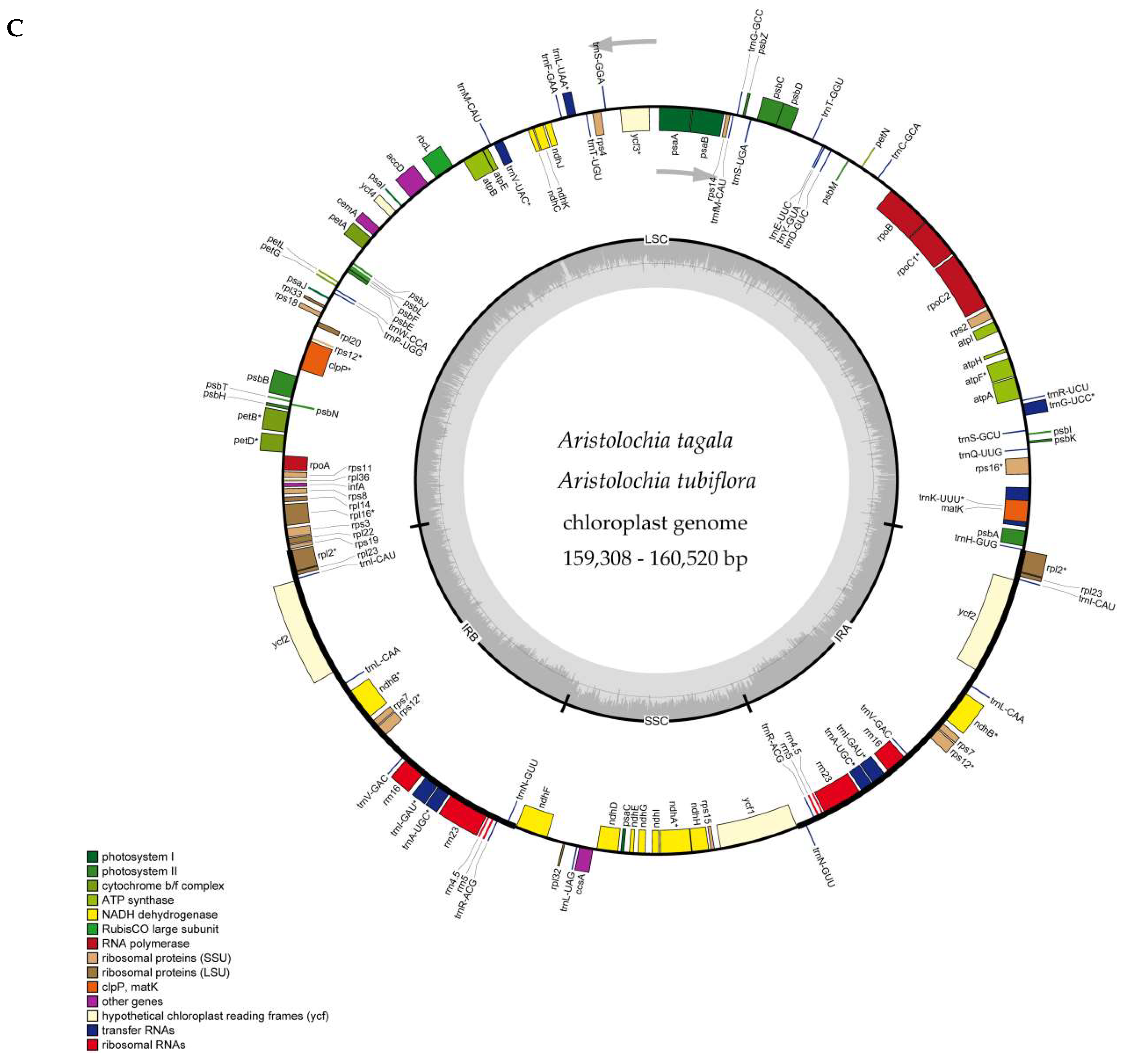
2.1. The Chloroplast Genome Structures of Species
2.2. IR Contraction and Expansion
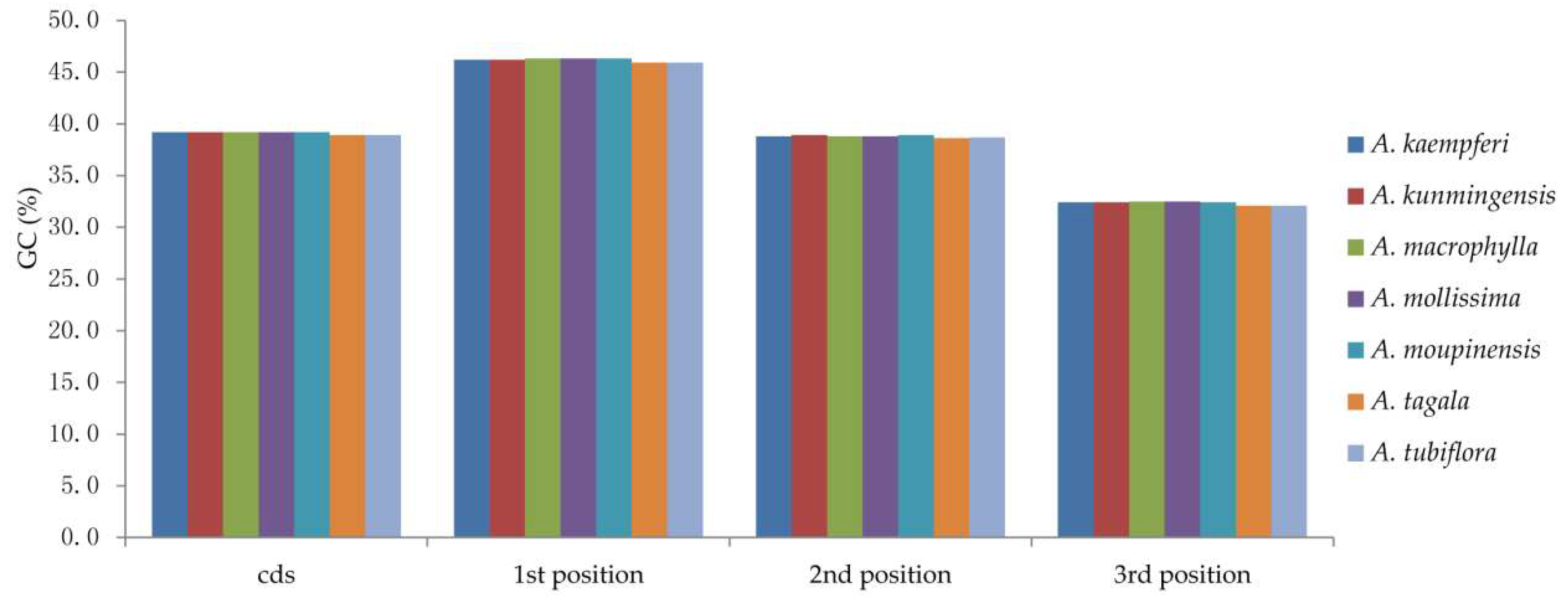
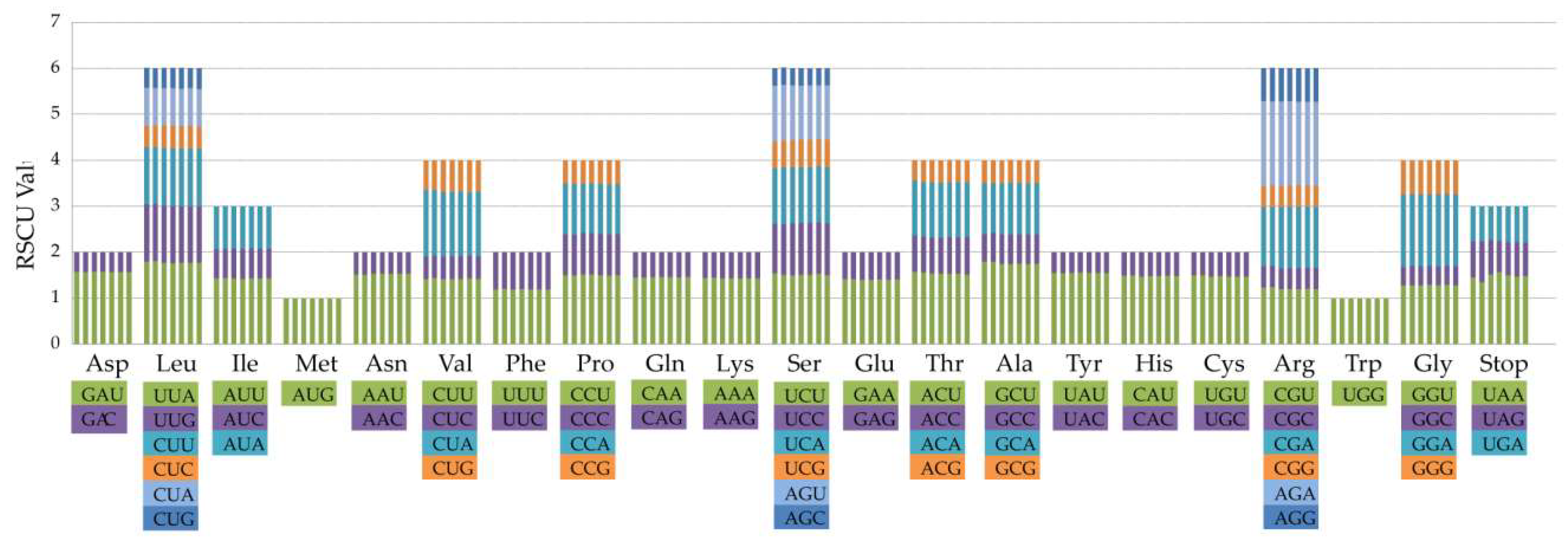
2.3. Codon Usage
2.4. Positive Selection Analysis
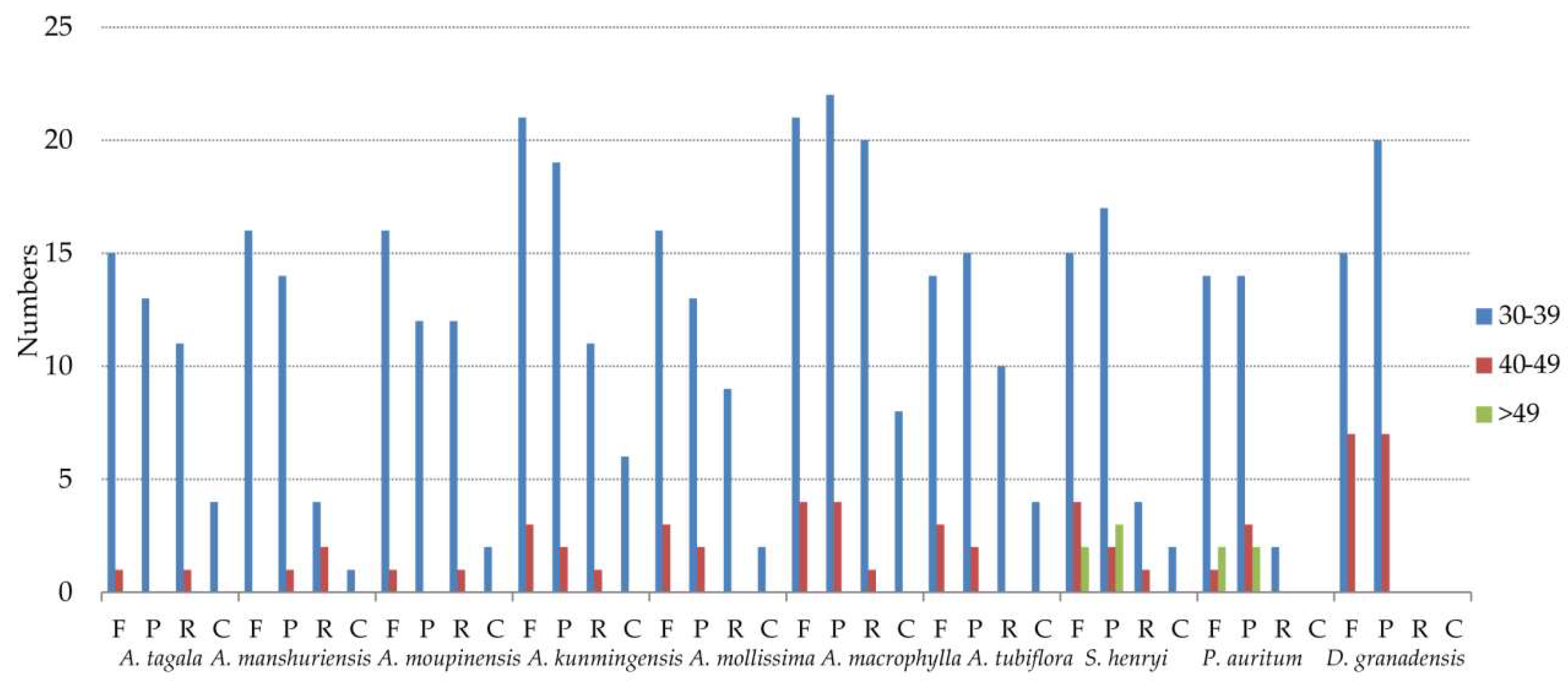
2.5. Repeat Structure and Simple Sequence Repeats Analyses
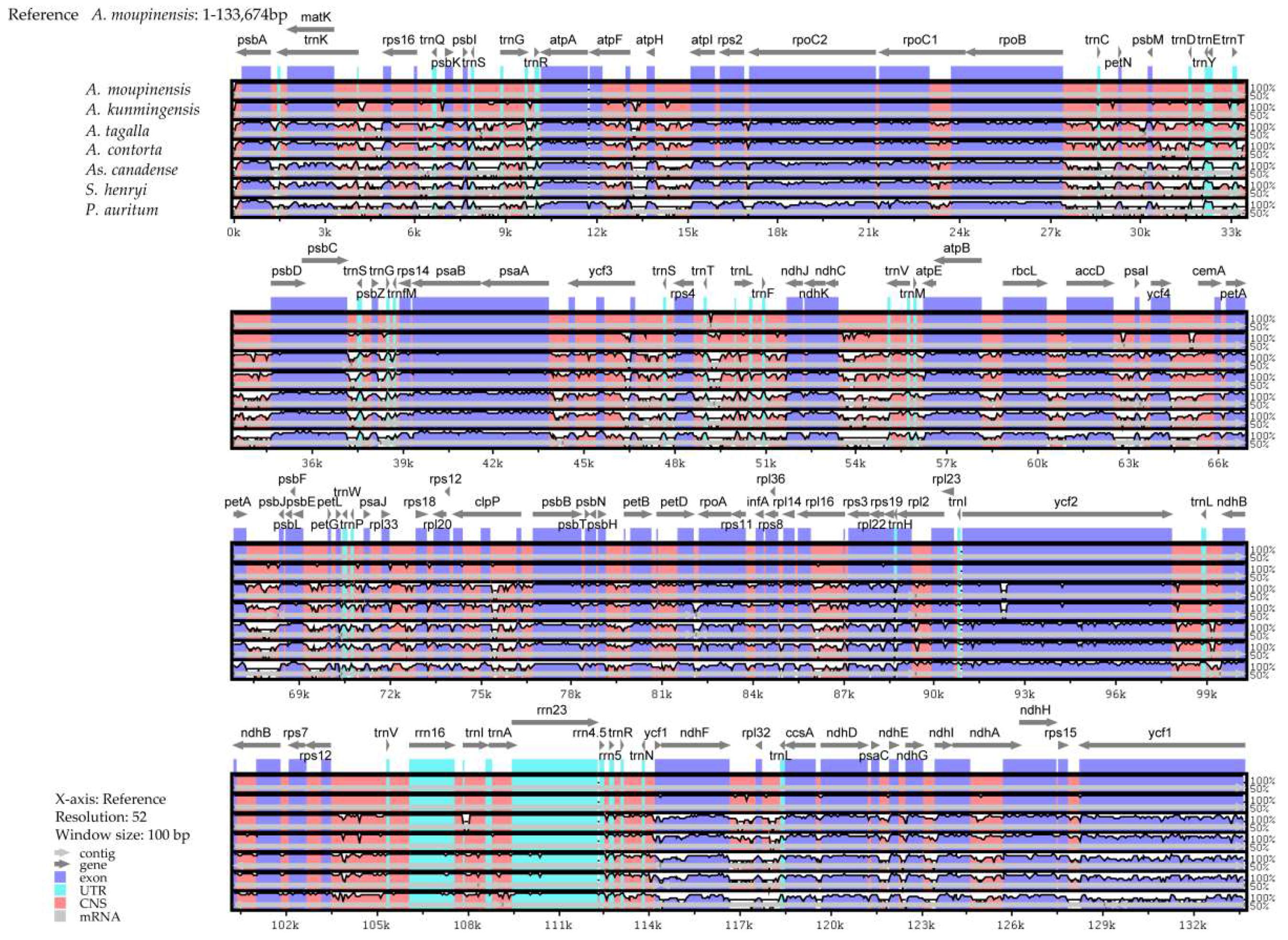
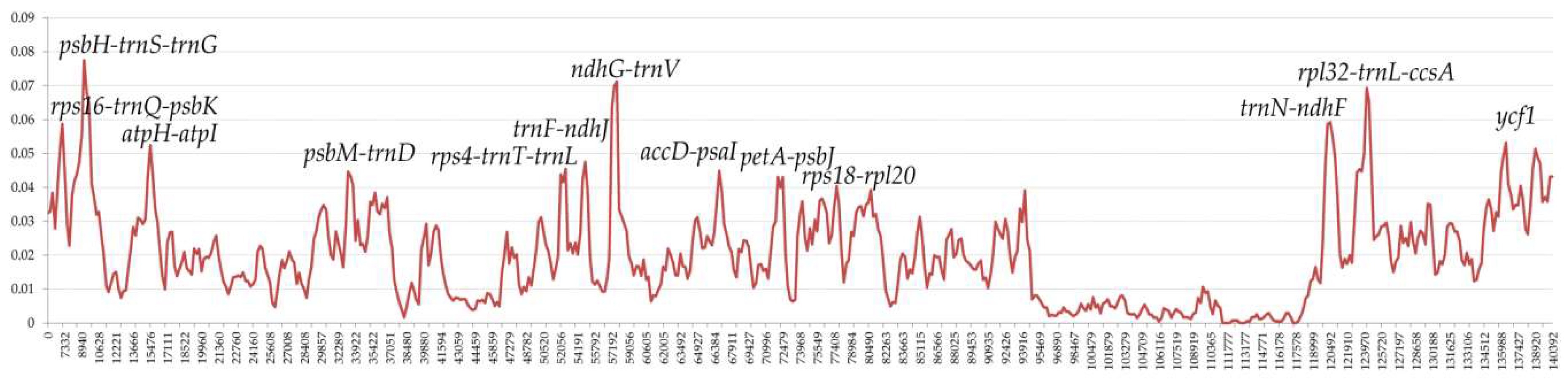
2.6. Comparative Genomic Divergence and Hotspots Regions
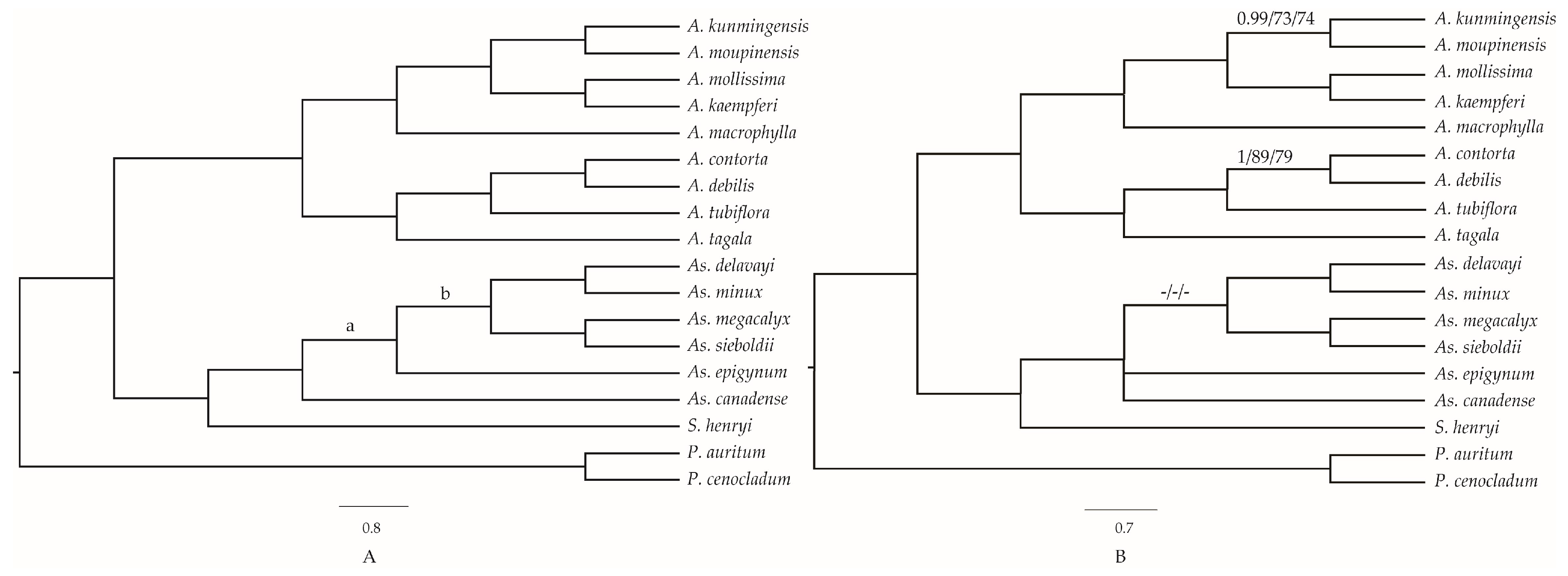
2.7. Phylogenetic Analyses
3. Discussion
3.1. IR Contraction and Expansion
3.2. Inferring the Phylogeny and Species Identification of Aristolochia
4. Materials and Methods
4.1. Plant Material, DNA Extraction, and Sequencing
4.2. Chloroplast Genome Assembly and Annotation
4.3. Genome Structure Analyses
4.4. Positive Selection Analysis
4.5. Genome Comparison and Nucleotide Variation Analysis
4.6. Phylogenetic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RSCU | Relative synonymous codon usage |
| NGS | Next-generation sequencing |
| TLA | Three letter acronym |
| SSR | Simple sequence repeats |
| ATP | Adenosine triphosphate |
| MP | Maximum parsimony |
| ML | Maximum likelihood |
| Pi | Nucleotide diversity |
| BI | Bayesian Inference |
| AAs | Aristolochic acids |
| LSC | Large single copy |
| SSC | Small single copy |
| CDS | Coding sequence |
| IR | Inverted repeat |
| SC | Single copy |
| CP | Chloroplast |
References
- Neinhuis, C.; Wanke, S.; Hilu, K.W.; Müller, K.; Borsch, T. Phylogeny of Aristolochiaceae based on parsimony, likelihood, and Bayesian analyses of trnL-trnF sequences. Plant Syst. Evol. 2005, 250, 7–26. [Google Scholar] [CrossRef]
- Wagner, S.T.; Isnard, S.; Rowe, N.P.; Samain, M.S.; Neinhuis, C.; Wanke, S. Escaping the lianoid habit: Evolution of shrub-like growth forms in Aristolochia subgenus Isotrema (Aristolochiaceae). Am. J. Bot. 2012, 99, 1609–1629. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.X.; Lin, S.Q.; Zhang, X.Q. Botanical and pharmacognostical studies of Chinese Aristolochia: Resource utilization. Acta Pharm. Sin. 1983, 18, 291–298. [Google Scholar]
- Ma, J.S. Medicinal resources of Aristolochia in China. Chin. Wild Plant Resour. 1988, 3, 31–34. [Google Scholar]
- Chen, C.H.; Dickman, K.G.; Moriya, M.; Zavadil, J.; Sidorenko, V.S.; Edwards, K.L.; Gnatenkod, D.V.; Wu, L.; Turesky, R.J.; Wu, X.R.; et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. USA 2012, 109, 8241–8246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Sun, W.; Wang, B.; Zhao, H.; Li, Y.; Cai, S.; Li, X.; Zhu, Y.J.; Yao, H.; Song, J.Y.; et al. An integrated system for identifying the hidden assassins in traditional medicines containing aristolochic acids. Sci. Rep. 2015, 5, 11318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, D.M.; Kang, J.J.; Lee, S.S.; Wang, S.Y.; Tsai, I.; Chen, G.Y.; Liao, H.W.; Chu, L.W.; Kuo, C.H.; Tseng, Y.J. Metabolomic analysis of complex Chinese remedies: Examples of induced nephrotoxicity in the mouse from a series of remedies containing aristolochic acid. Evid.-Based Complement. Altern. Med. 2013, 2013, 263757. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.W.T.; Poon, S.L.; Huang, M.N.; Lim, J.Q.; Boot, A.; Yu, W.; Suzuki, Y.; Thangaraju, S.; Ng, C.C.Y.; Tan, P.; et al. Aristolochic acids and their derivatives are widely implicated in liver cancers in Taiwan and throughout Asia. Sci. Transl. Med. 2017, 9, eaan6446. [Google Scholar] [CrossRef] [PubMed]
- Martena, M.J.; Van der Wielen, J.C.A.; Van de Laak, L.F.J.; Konings, E.J.M.; de Groot, H.N.; Rietjens, I.M.C.M. Enforcement of the ban on aristolochic acids in chinese traditional herbal preparations on the dutch market. Anal. Bioanal. Chem. 2007, 389, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.K.; González, F.A. Phylogenetic Relationships in Aristolochiaceae. Syst. Bot. 2003, 28, 236–249. [Google Scholar] [CrossRef]
- Wanke, S.; Jaramillo, M.A.; Borsch, T.; Samain, M.S.; Quandt, D.; Neinhuis, C. Evolution of Piperales-matK gene and trnK intron sequence data reveal lineage specific resolution contrast. Mol. Phylogenet. Evol. 2007, 42, 477–497. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.K. A cladistic analysis of Asarum and implications for the evolution of Herkogamy. Am. J. Bot. 1997, 84, 1752–1765. [Google Scholar]
- Lawrence, M.K. Phylogenetic relationships in Asarum (Aristolochiaceae) based on morphology and ITS sequences. Am. J. Bot. 1998, 85, 1454–1467. [Google Scholar]
- González, F.A.; Stevenson, D.W. A phylogenetic analysis of the subfamily Aristolochioideae (Aristolochiaceae). Rev. Acad. Colomb. Cienc. 2002, 66, 59–65. [Google Scholar]
- Ohi-Toma, T.; Sugawara, T.; Neihuis, C.; Murata, H.; Wanke, S.; Murata, J. Molecular phylogeny of Aristolochia sensu lato(Aristolochiaceae) based on sequences of rbcL, matK, and phyA genes, with special reference to differentiation of chromosome numbers. Syst. Bot. 2006, 31, 481–492. [Google Scholar] [CrossRef]
- Wanke, S.; Samain, M.S.; Vanderschaeve, L.; Mathieu, G.; Goetghebeur, P.; Neinhuis, C. Phylogeny of the genus Peperomia (Piperaceae) inferred from the trnK/matK region (cpDNA). Plant Biol. 2006, 8, 93–102. [Google Scholar] [CrossRef] [PubMed]
- González, F.; Wagner, S.T.; Salomo, K.; Symmank, L.; Samain, M.-S.; Isnard, S.; Rowe, N.P.; Neinhuis, C.; Wanke, S.; Carine, M. Present trans-Pacific disjunct distribution of Aristolochia subgenus Isotrema (Aristolochiaceae) was shaped by dispersal, vicariance and extinction. J. Biogeogr. 2014, 41, 380–391. [Google Scholar] [CrossRef]
- Dechbumroong, P.; Aumnouypol, S.; Denduangboripant, J.; Sukrong, S. DNA barcoding of Aristolochia plants and development of species-specific multiplex PCR to aid HPTLC in ascertainment of Aristolochia herbal materials. PLoS ONE 2018, 13, e0202625. [Google Scholar] [CrossRef] [PubMed]
- Sarma, B.; Tanti, B. Analysis of genetic diversity of certain species of Aristolochia using ISSR-based molecular markers. Curr. Life Sci. 2017, 3, 47–53. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Yi, T.S.; Zeng, L.Q.; Gong, X. The population genetic structure and diversification of Aristolochia delavayi (Aristolochiaceae), an endangered species of the dry hot valleys of the Jinsha River, southwestern China. Botany 2014, 92, 579–587. [Google Scholar] [CrossRef]
- Douglas, S.E. Plastid evolution: Origins, diversity, trends. Curr. Opin. Genet. Dev. 1998, 8, 655–661. [Google Scholar] [CrossRef]
- Moore, M.J.; Bell, C.D.; Soltis, P.S.; Soltis, D.E. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Am. Sci. USA 2007, 104, 19363–19368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.B.; Yang, S.X.; Li, H.T.; Jing, Y.; Li, D.Z. Comparative chloroplast genomes of Camellia species. PLoS ONE 2013, 8, e73053. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.; Gao, L. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Boil. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Ni, D.; Wang, Y.; Shao, J.; Wang, X.; Yang, D.; Wang, J.S.; Chen, H.M.; Liu, C. Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus Membranaceus. Sci. Rep. 2016, 6, 21669. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Raubeson, L.A.; Boore, J.L.; Depamphilis, C.W.; Chumley, T.W.; Haberle, R.C. Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 2005, 395, 348–384. [Google Scholar] [PubMed]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.G.; Chen, X.L.; Cui, Y.X.; Sun, W.; Li, Y.H.; Wang, Y.; Song, J.Y.; Yao, H. Molecular Structure and Phylogenetic Analyses of Complete Chloroplast Genomes of Two Aristolochia Medicinal Species. Int. J. Mol. Sci. 2017, 18, 1839. [Google Scholar] [CrossRef] [PubMed]
- Sinn, B.T.; Sedmak, D.D.; Kelly, L.M.; Freudenstein, J.V. Total duplication of the small single copy region in the angiosperm plastome: Rearrangement and inverted repeat instability in Asarum. Am. J. Bot. 2018, 105, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Sakaguchi, S.; Isagi, Y.; Setoguchi, H. Comparative chloroplast genomics of series Sakawanum in genus Asarum (Aristolochiaceae) to develop single nucleotide polymorphisms (SNPs) and simple sequence repeat (SSR) markers. J. For. Res. 2018, 23, 387–392. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Ueno, S.; Tsumura, Y. Application of a simplified method of chloroplast enrichment to small amounts of tissue for chloroplast genome sequencing. Appl. Plant Sci. 2017, 5, 1700002. [Google Scholar] [CrossRef] [PubMed]
- NCBI. Genome. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 14 January 2019).
- Xu, J.W.; Feng, D.J.; Song, G.S.; Wei, X.L.; Chen, L.; Wu, X.L.; Li, X.G.; Zhu, Z. The first intron of rice epsp synthase enhances expression of foreign gene. Sci. China Life Sci. 2003, 46, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus Macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.J.; Al-Mssallem, I.S.; Yu, J. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean Ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Boil. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Huotari, T.; Korpelainen, H. Complete chloroplast genome sequence of Elodea canadensis and comparative analyses with other monocot plastid genomes. Gene 2012, 508, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ma, P.F.; Li, H.T.; Yang, J.B.; Wang, H.; Li, D.Z. Plastid phylogenomic analyses resolve Tofieldiaceae as the root of the early diverging monocot order Alismatales. Genome Biol. Evol. 2016, 8, 932–945. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.Q.; Penaflor, C.; Kuehl, J.V.; Leebens-Mack, J.; Carlson, J.E.; Pamphilis, C.W.D.; Boore, J.L.; Jansen, R.K. Complete plastid genome sequences of Drimys, Liriodendron, and Piper: Implications for the phylogenetic relationships of magnoliids. BMC Evol. Boil. 2006, 6, 77. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, H.C.; Lin, I.; Chow, T.Y.; Chen, H.H.; Chen, W.H.; Cheng, C.H.; Lin, C.Y.; Liu, S.M.; Chang, C.C.; et al. The chloroplast genome of Phalaenopsis aphrodite (Orchidaceae): Comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications. Mol. Biol. Evol. 2006, 23, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Borsch, T.; Quandt, D. Mutational dynamics and phylogenetic utility of noncoding chloroplast DNA. Plant Syst. Evol. 2009, 282, 169–199. [Google Scholar] [CrossRef]
- Dong, W.P.; Liu, J.; Yu, J.; Wang, L.; Zhou, S.L. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Kim, T.S.; Park, Y.J. Rice chloroplast genome variation architecture and phylogenetic dissection in diverse Oryza species assessed by whole-genome resequencing. Rice 2016, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Liu, H.; Xu, C.; Zuo, Y.J.; Chen, Z.J.; Zhou, S.L. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.P.; Bi, Y.; Yang, F.P.; Zhang, M.F.; Chen, X.Q.; Xue, J.; Zhang, X.H. Complete chloroplast genome sequences of Lilium: Insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 2017, 7, 5751. [Google Scholar] [CrossRef] [PubMed]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.J.; Lv, S.Z.; Zhang, Y.X.; Du, X.H.; Wang, L.; Biradar, S.S.; Tan, X.F.; Wan, F.H.; Song, W.N. Complete chloroplast genome sequence of a major invasive species, crofton weed (Ageratina adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, R.H.; Li, X.G. Comparative analysis of the complete chloroplast genome of four known Ziziphus species. Genes 2017, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.H.; Tembrock, L.R.; Zheng, S.Y.; Wu, Z.Q. The complete chloroplast genome of Catha edulis: A comparative analysis of genome features with related species. Int. J. Mol. Sci. 2018, 19, 525. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.; Morgante, M.; Mcdevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.P.; Xu, C.; Li, C.H.; Sun, J.H.; Zuo, Y.J.; Shi, S.; Cheng, T.; Guo, J.J.; Zhou, S.L. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Muto, A.; Ushida, C. Transcription and translation. Methods Cell Biol. 1995, 48, 483. [Google Scholar]
- Koren, O.G.; Nakonechnaya, O.V.; Zhuravlev, Y.N. Genetic structure of natural populations of the relict species Aristolochia manshuriensis (Aristolochiaceae) in disturbed and intact habitats. Russ. J. Genet. 2009, 45, 678–684. [Google Scholar] [CrossRef]
- Nakonechnaya, O.V.; Kholina, A.B.; Koren, O.G.; Zhuravlev, Y.N. Genetic diversity of a rare species Aristolochia contorta Bunge (Aristolochiaceae) in Primorsky krai. Russ. J. Genet. 2012, 48, 152–162. [Google Scholar] [CrossRef]
- Watanabe, K.; Kajita, T.; Murata, J. Chloroplast DNA and variation and geographical structure of the Aristolochia kaempferi group (Aristolochiaceae). Am. J. Bot. 2006, 93, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Ohi-Toma, T.; Murata, J. Multiple hybridization in the Aristolochia kaempferi group (Aristolochiaceae): Evidence from reproductive isolation and molecular phylogeny. Am. J. Bot. 2008, 95, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Ashton, B.; Buxton, S.; Cheung, M.; Cooper, A.; Duran, C.; Field, M. Geneious; Version 5.3+; Biomatters: Auckland, New Zealand, 2011. [Google Scholar]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organelle genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbrichtjones, E.S.; Fischer, A.; Bock, R.; Greiner, S. Geseq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Shi, L.C.; Zhu, Y.; Chen, H.M.; Zhang, J.H.; Lin, X.H.; Guan, X.J. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; Mcquillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. Organellargenomedraw (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Koichiro, T.; Glen, S.; Daniel, P.; Alan, F.; Sudhir, K. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. Reputer: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.W.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. Vista: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, w273–w279. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. Bioedit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Rozas, J.; Sánchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. Dnasp, dna polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony, Distance, and Likelihood; Version 4; Sinauer: Sunderland, MA, USA, 2002. [Google Scholar]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. GCE 2010, 14, 1–8. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 10, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Bui, M.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Figtree v1.4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 14 January 2019).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Total | LSC | IR | SSC | CDS | Total | Protein Coding Genes | tRNA | rRNA | GC% |
|---|---|---|---|---|---|---|---|---|---|---|
| A. kaempferi | 159,612 | 88,890 | 25,681 | 19,360 | 79191.0 | 113 | 79 | 30 | 4 | 38.8 |
| A. kunmingensis | 160,051 | 89,308 | 25,698 | 19,347 | 79143.0 | 113 | 79 | 30 | 4 | 38.7 |
| A. macrophylla | 160,493 | 89,788 | 25,664 | 19,377 | 79116.0 | 113 | 79 | 30 | 4 | 38.6 |
| A. mollissima | 159,653 | 88,948 | 25,681 | 19,338 | 79194.0 | 113 | 79 | 30 | 4 | 38.8 |
| A. moupinensis | 159,374 | 88,652 | 25,700 | 19,322 | 78753.0 | 113 | 79 | 30 | 4 | 38.7 |
| A. tagala | 159,308 | 89,414 | 25,242 | 19,410 | 78582.0 | 113 | 79 | 30 | 4 | 38.5 |
| A. tubiflora | 160,520 | 89,859 | 25,431 | 19,799 | 78624.0 | 113 | 79 | 30 | 4 | 38.8 |
| No. | Group of Genes | Genes Names | Amount |
|---|---|---|---|
| 1 | Photosystems I | psaA, psaB, psaC, psaI, psaJ | 5 |
| 2 | Photosystems II | psbA, psbB, psbC, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 |
| 3 | Cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | 6 |
| 4 | ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | 6 |
| 5 | NADH dehydrogenase | ndhA *, ndhB *(x2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 12 (1) |
| 6 | Rubisco large subunit | rbcL | 1 |
| 7 | RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | 4 |
| 8 | Ribosomal proteins(SSU) | rps2, rps3, rps7(x2), rps8, rps11, rps12 **(x2), rps14, rps15, rps16 *, rps18, rps19 | 14 (2) |
| 9 | Ribosomal proteins(LSU) | rpl2 *(x2), rpl14, rpl16 *, rpl20, rpl22, rpl23(x2), rpl32, rpl33, rpl36 | 11 (2) |
| 10 | Assembly/stability of photosystem I | ycf3 **, ycf4 | 2 |
| 11 | Transfer RNAs | 37/38 tRNAs (6 contain an intron, 7/8 in the IRs) | 37 (7)/38(8) |
| 12 | Ribosomal RNAs | rrn4.5(x2), rrn5(x2), rrn16(x2), rrn23(x2) | 8 (8) |
| 13 | RNA processing | matK | 1 |
| 14 | Carbon metabolism | cemA | 1 |
| 15 | Cytochrome c synthesis | ccsA | 1 |
| 16 | Proteins of unknown function | ycf1, ycf2(x2) | 3 (1) |
| 17 | Other genes | accD, clpP **, infA | 3 |
| Taxon | Gene | Location | Exon I | Intron I | Exon II | Intron II | Exon III |
|---|---|---|---|---|---|---|---|
| A. kunmingensis | atpF | LSC | 144 | 792 | 411 | ||
| clpP | LSC | 71 | 912 | 288 | 674 | 250 | |
| ndhA | SSC | 551 | 1095 | 541 | |||
| ndhB | IR | 777 | 703 | 756 | |||
| petB | LSC | 6 | 215 | 642 | |||
| petD | LSC | 6 | 702 | 477 | |||
| rpl16 | LSC | 9 | 1092 | 402 | |||
| rpl2 | IR | 391 | 700 | 431 | |||
| rpoC1 | LSC | 432 | 762 | 1617 | |||
| rps12 | LSC | 114 | 232 | 536 | 23 | ||
| rps16 | LSC | 46 | 842 | 221 | |||
| trnA-UGC | IR | 38 | 804 | 35 | |||
| trnG-UCC | LSC | 24 | 763 | 48 | |||
| trnI-GAU | IR | 37 | 936 | 35 | |||
| trnK-UUU | LSC | 37 | 2574 | 35 | |||
| trnL-UAA | LSC | 35 | 454 | 50 | |||
| trnV-UAC | LSC | 37 | 587 | 36 | |||
| ycf3 | LSC | 126 | 871 | 226 | 745 | 155 | |
| A. mollissima | atpF | LSC | 144 | 772 | 411 | ||
| clpP | LSC | 71 | 897 | 295 | 672 | 243 | |
| ndhA | SSC | 551 | 1097 | 541 | |||
| ndhB | IR | 777 | 702 | 756 | |||
| petB | LSC | 6 | 215 | 642 | |||
| petD | LSC | 6 | 702 | 477 | |||
| rpl16 | LSC | 9 | 1098 | 399 | |||
| rpl2 | IR | 391 | 700 | 431 | |||
| rpoC1 | LSC | 432 | 762 | 1617 | |||
| rps12 | LSC | 114 | 232 | 536 | 23 | ||
| rps16 | LSC | 46 | 848 | 221 | |||
| trnA-UGC | IR | 38 | 804 | 35 | |||
| trnG-UCC | LSC | 24 | 764 | 47 | |||
| trnI-GAU | IR | 37 | 936 | 35 | |||
| trnK-UUU | LSC | 37 | 2562 | 35 | |||
| trnL-UAA | LSC | 35 | 455 | 50 | |||
| trnV-UAC | LSC | 37 | 587 | 36 | |||
| ycf3 | LSC | 126 | 820 | 226 | 753 | 155 | |
| A. kaempferi | atpF | LSC | 144 | 780 | 411 | ||
| clpP | LSC | 71 | 900 | 295 | 671 | 243 | |
| ndhA | SSC | 551 | 1097 | 541 | |||
| ndhB | IR | 777 | 702 | 756 | |||
| petB | LSC | 6 | 215 | 642 | |||
| petD | LSC | 6 | 702 | 477 | |||
| rpl16 | LSC | 9 | 1101 | 399 | |||
| rpl2 | IR | 391 | 700 | 431 | |||
| rpoC1 | LSC | 432 | 765 | 1617 | |||
| rps12 | LSC | 114 | 232 | 536 | 23 | ||
| rps16 | LSC | 46 | 842 | 221 | |||
| trnA-UGC | IR | 38 | 804 | 35 | |||
| trnG-UCC | LSC | 24 | 764 | 48 | |||
| trnI-GAU | IR | 37 | 936 | 35 | |||
| trnK-UUU | LSC | 37 | 2552 | 35 | |||
| trnL-UAA | LSC | 50 | 455 | 35 | |||
| trnV-UAC | LSC | 37 | 587 | 36 | |||
| ycf3 | LSC | 126 | 812 | 226 | 752 | 155 | |
| A. moupinensis | atpF | LSC | 144 | 789 | 411 | ||
| clpP | LSC | 71 | 909 | 288 | 669 | 250 | |
| ndhA | SSC | 551 | 1101 | 541 | |||
| ndhB | IR | 777 | 703 | 756 | |||
| petB | LSC | 6 | 211 | 646 | |||
| petD | LSC | 6 | 708 | 477 | |||
| rpl16 | LSC | 399 | 1100 | 9 | |||
| rpl2 | IR | 391 | 700 | 431 | |||
| rpoC1 | LSC | 432 | 764 | 1617 | |||
| rps12 | LSC | 114 | 232 | 536 | 23 | ||
| rps16 | LSC | 46 | 839 | 221 | |||
| trnA-UGC | IR | 38 | 804 | 35 | |||
| trnG-UCC | LSC | 24 | 758 | 48 | |||
| trnI-GAU | IR | 37 | 936 | 35 | |||
| trnK-UUU | LSC | 37 | 2567 | 35 | |||
| trnL-UAA | LSC | 35 | 462 | 50 | |||
| trnV-UAC | LSC | 37 | 587 | 36 | |||
| ycf3 | LSC | 126 | 920 | 226 | 746 | 155 | |
| A. macrophylla | atpF | LSC | 144 | 778 | 411 | ||
| clpP | LSC | 71 | 928 | 288 | 664 | 250 | |
| ndhA | SSC | 551 | 1084 | 541 | |||
| ndhB | IR | 777 | 702 | 756 | |||
| petB | LSC | 6 | 215 | 642 | |||
| petD | LSC | 6 | 706 | 477 | |||
| rpl16 | LSC | 9 | 1095 | 402 | |||
| rpl2 | IR | 391 | 700 | 431 | |||
| rpoC1 | LSC | 432 | 788 | 1617 | |||
| rps12 | LSC | 114 | 232 | 536 | 23 | ||
| rps16 | LSC | 46 | 836 | 221 | |||
| trnA-UGC | IR | 38 | 804 | 35 | |||
| trnG-UCC | LSC | 24 | 755 | 47 | |||
| trnI-GAU | IR | 37 | 936 | 35 | |||
| trnK-UUU | LSC | 37 | 2558 | 35 | |||
| trnL-UAA | LSC | 35 | 475 | 50 | |||
| trnV-UAC | LSC | 37 | 589 | 36 | |||
| ycf3 | LSC | 126 | 892 | 226 | 757 | 155 | |
| A.tubiflora | atpF | LSC | 144 | 751 | 411 | ||
| clpP | LSC | 71 | 819 | 288 | 671 | 250 | |
| ndhA | SSC | 551 | 1079 | 541 | |||
| ndhB | IR | 777 | 705 | 756 | |||
| petB | LSC | 6 | 214 | 642 | |||
| petD | LSC | 6 | 693 | 477 | |||
| rpl16 | LSC | 9 | 1077 | 399 | |||
| rpl2 | IR | 391 | 657 | 431 | |||
| rpoC1 | LSC | 432 | 780 | 1617 | |||
| rps12 | LSC | 114 | 232 | 536 | 23 | ||
| rps16 | LSC | 46 | 889 | 221 | |||
| trnA-UGC | IR | 38 | 809 | 35 | |||
| trnG-UCC | LSC | 24 | 768 | 47 | |||
| trnI-GAU | IR | 37 | 937 | 35 | |||
| trnK-UUU | LSC | 37 | 2635 | 35 | |||
| trnL-UAA | LSC | 35 | 514 | 50 | |||
| trnV-UAC | LSC | 37 | 594 | 36 | |||
| ycf3 | LSC | 126 | 764 | 226 | 752 | 149 | |
| A. tagala | atpF | LSC | 144 | 778 | 408 | ||
| clpP | LSC | 71 | 802 | 288 | 671 | 250 | |
| ndhA | SSC | 551 | 1101 | 541 | |||
| ndhB | IR | 777 | 704 | 756 | |||
| petB | LSC | 6 | 219 | 642 | |||
| petD | LSC | 6 | 488 | 477 | |||
| rpl16 | LSC | 9 | 1071 | 399 | |||
| rpl2 | IR | 391 | 657 | 431 | |||
| rpoC1 | LSC | 432 | 785 | 1617 | |||
| rps12 | LSC | 114 | 232 | 536 | 23 | ||
| rps16 | LSC | 46 | 848 | 221 | |||
| trnA-UGC | IR | 38 | 804 | 35 | |||
| trnG-UCC | LSC | 24 | 768 | 48 | |||
| trnI-GAU | IR | 37 | 743 | 35 | |||
| trnK-UUU | LSC | 37 | 2687 | 35 | |||
| trnL-UAA | LSC | 35 | 490 | 50 | |||
| trnV-UAC | LSC | 37 | 595 | 36 | |||
| ycf3 | LSC | 126 | 830 | 226 | 763 | 149 |
| Gene name | M2a | M8 | ||
|---|---|---|---|---|
| selected sites | Pr (w > 1) | selected sites | Pr (w > 1) | |
| rpl20 | 71A | 0.918 | 71A | 0.967 * |
| 72L | 0.999 ** | 72L | 1.000 ** | |
| 105R | 0.963 * | 105R | 0.984 * | |
| 116H | 0.963 * | 116H | 0.988 * | |
| rps12 | 79M | 0.966 * | 79M | 0.987 * |
| rps18 | 4S | 0.937 | 4S | 0.975 * |
| 99T | 0.921 | 99T | 0.967 * | |
| ycf1 | 206S | 0.914 | 206S | 0.967 * |
| 211V | 0.975 * | 211V | 0.989 * | |
| 1412N | 0.922 | 1412N | 0.971 * | |
| ycf2 | 2036W | 0.932 | 2036W | 0.950 * |
| Regions | Number of Sites | Variable Sites | Parsimony Informative Sites | Nucleotide Diversity |
|---|---|---|---|---|
| LSC | 94,564 | 4430 | 2315 | 0.02182 |
| SSC | 20,451 | 1433 | 804 | 0.03114 |
| IR | 25,884 | 253 | 154 | 0.00411 |
| Complete | 166,113 | 6422 | 3461 | 0.01717 |
| CDS | 79,365 | 2528 | 1376 | 0.01337 |
| High Variable Marker | Length | Variable Sites | Parsimony Informative Sites | Nucleotide Diversity |
|---|---|---|---|---|
| rps16-trnQ-psbK | 1301 | 104 | 58 | 0.04278 |
| psbI-trnS-trnG | 2357 | 257 | 159 | 0.05364 |
| atpH-atpI | 1160 | 104 | 69 | 0.04439 |
| psbM-trnD | 1119 | 152 | 75 | 0.05888 |
| rps4-trnT-trnL | 1572 | 105 | 50 | 0.04178 |
| trnF-ndhJ | 920 | 85 | 51 | 0.04216 |
| ndhC-trnV | 1402 | 152 | 97 | 0.06311 |
| accD-psaI | 637 | 61 | 35 | 0.04220 |
| petA-psbJ | 1130 | 106 | 65 | 0.04444 |
| rps18-rpl20 | 682 | 58 | 36 | 0.04155 |
| trnN-ndhF | 1492 | 161 | 86 | 0.04758 |
| rpl32-trnL-ccsA | 2679 | 202 | 113 | 0.04608 |
| ycf1a | 1225 | 126 | 74 | 0.04285 |
| ycf1b | 652 | 56 | 32 | 0.04053 |
| ycf1c | 1228 | 134 | 84 | 0.04611 |
| ycf1d | 740 | 70 | 39 | 0.04278 |
| Combine | 20296 | 2216 | 1349 | 0.05413 |
| Species | Samples | Voucher | locality |
|---|---|---|---|
| A. kaempferi | E2265 | Yuan Wang | Japan, Tokyo |
| A. kunmingensis | E754 | Zhanghua Wang | China, Yunnan |
| A. macrophylla | E2111 | Jinshuang Ma | North America, North Carolia |
| A. mollissima | E1016 | Xinxin Zhu & Zhixiang Hua | China, Guangdong |
| A. moupinensis | E1086 | Xinxin Zhu & Zhixiang Hua | China, Sichuan |
| A. tagala | E1071 | Yuan Wang | China, Hongkong |
| A. tubiflora | E2239 | Shuwan Li | China, Guangxi |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Zuo, Y.; Zhu, X.; Liao, S.; Ma, J. Complete Chloroplast Genomes and Comparative Analysis of Sequences Evolution among Seven Aristolochia (Aristolochiaceae) Medicinal Species. Int. J. Mol. Sci. 2019, 20, 1045. https://doi.org/10.3390/ijms20051045
Li X, Zuo Y, Zhu X, Liao S, Ma J. Complete Chloroplast Genomes and Comparative Analysis of Sequences Evolution among Seven Aristolochia (Aristolochiaceae) Medicinal Species. International Journal of Molecular Sciences. 2019; 20(5):1045. https://doi.org/10.3390/ijms20051045
Chicago/Turabian StyleLi, Xiaoqin, Yunjuan Zuo, Xinxin Zhu, Shuai Liao, and Jinshuang Ma. 2019. "Complete Chloroplast Genomes and Comparative Analysis of Sequences Evolution among Seven Aristolochia (Aristolochiaceae) Medicinal Species" International Journal of Molecular Sciences 20, no. 5: 1045. https://doi.org/10.3390/ijms20051045