Comparative Analyses of Five Complete Chloroplast Genomes from the Genus Pterocarpus (Fabacaeae)


Abstract
:1. Introduction
2. Results
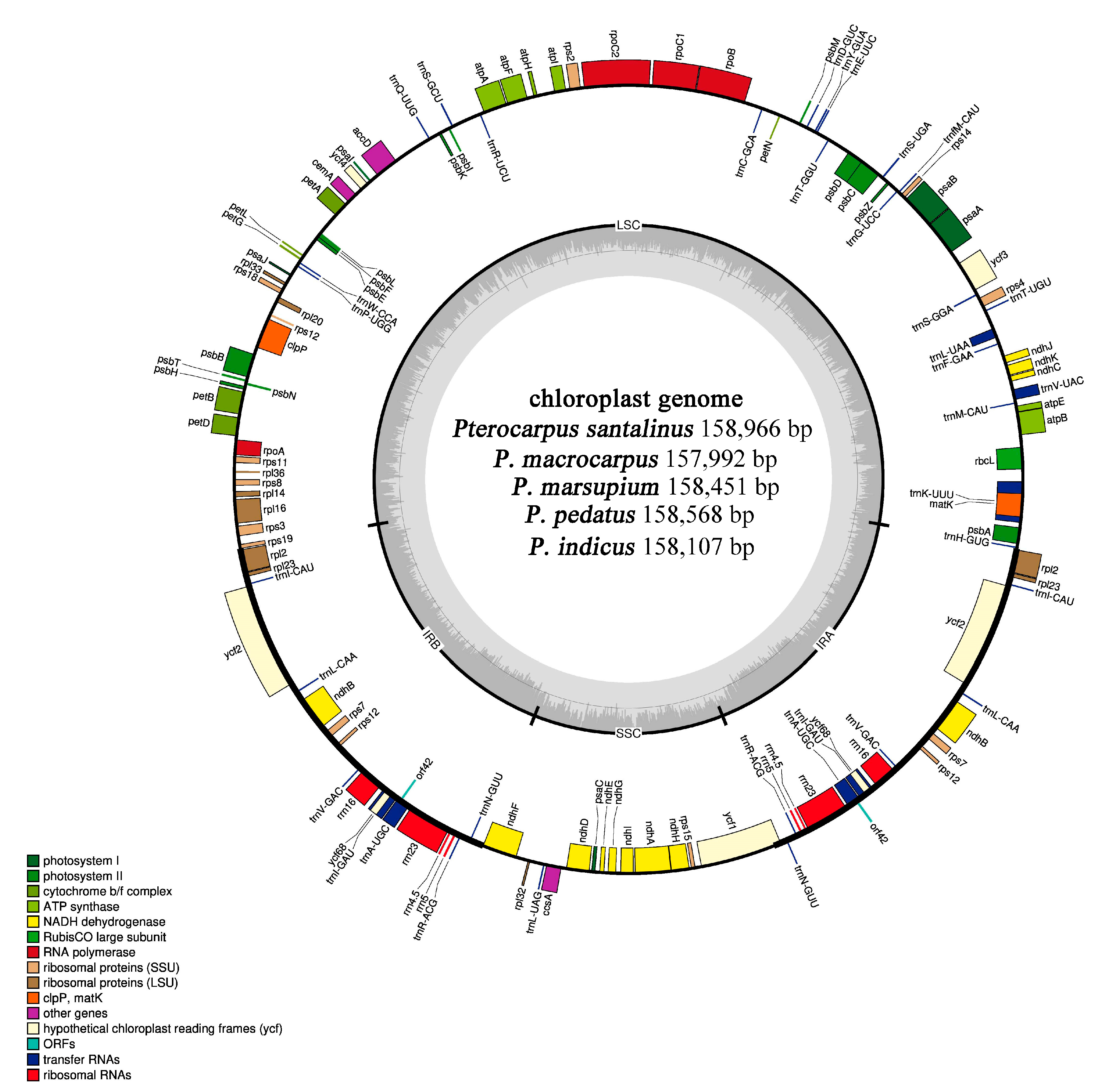
2.1. Genomic Characteristics of Chloroplast
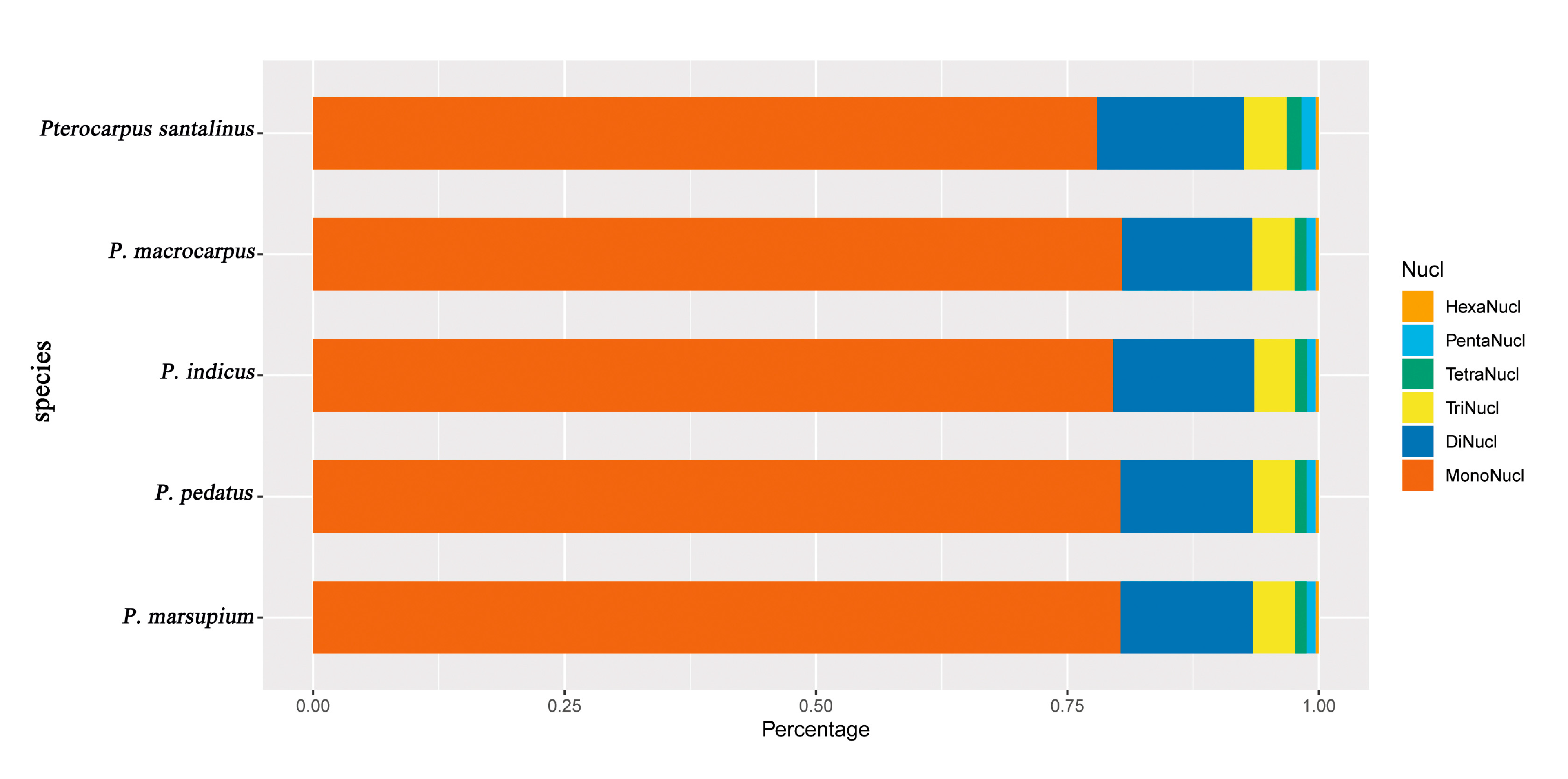
2.2. Detection of Chloroplast Repeat Sequences and SSRs
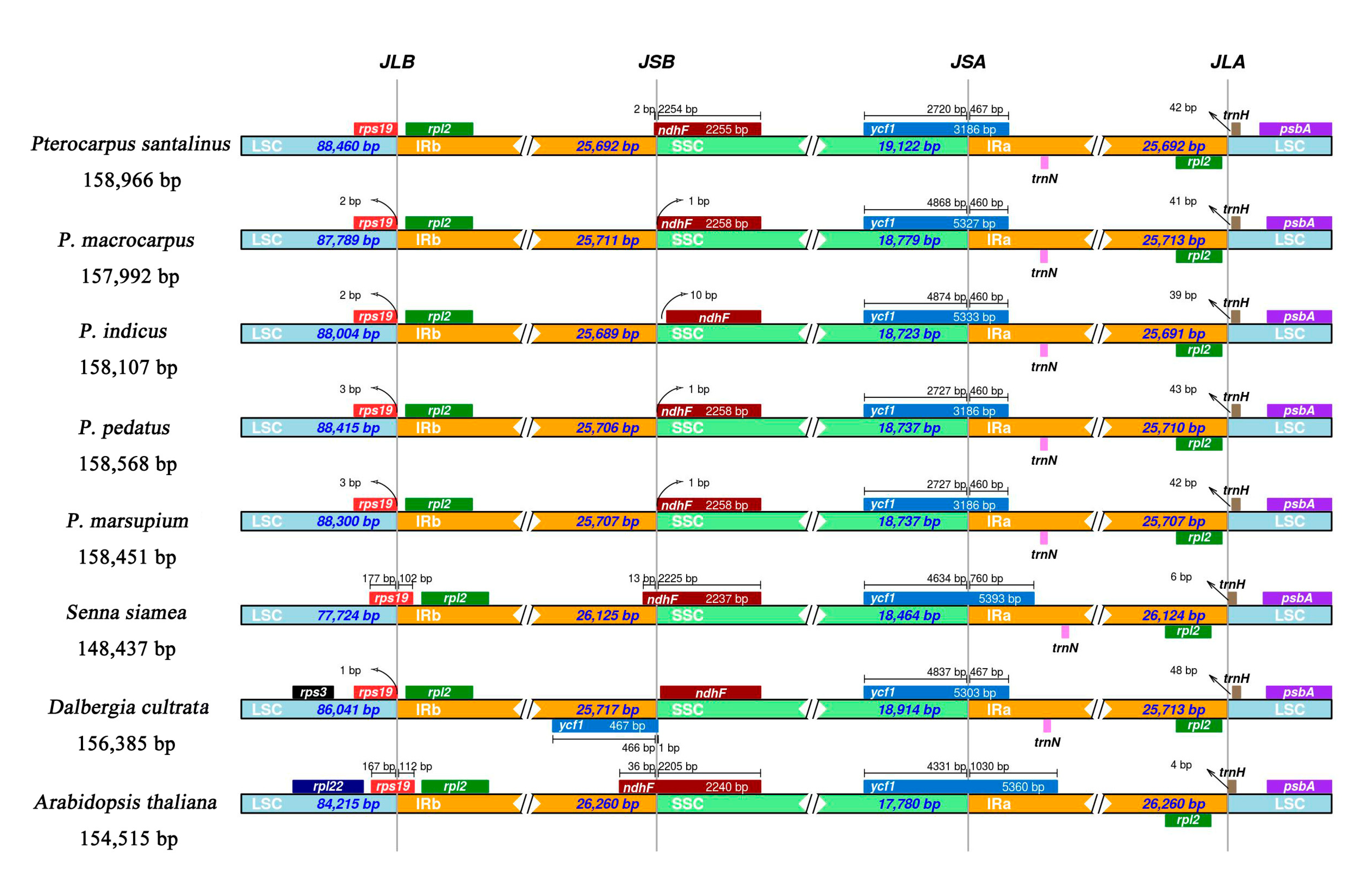
2.3. IR Expansion and Contraction
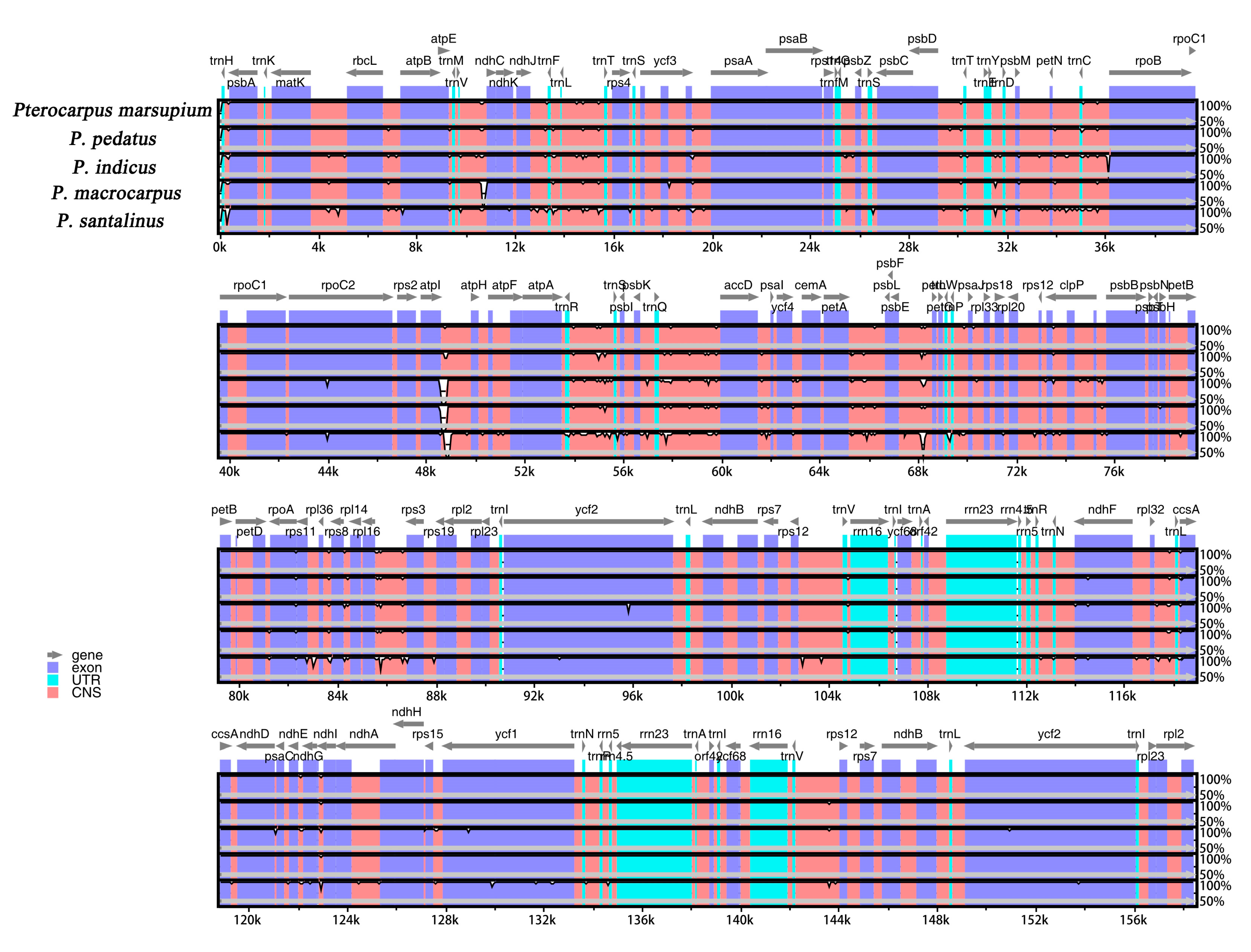
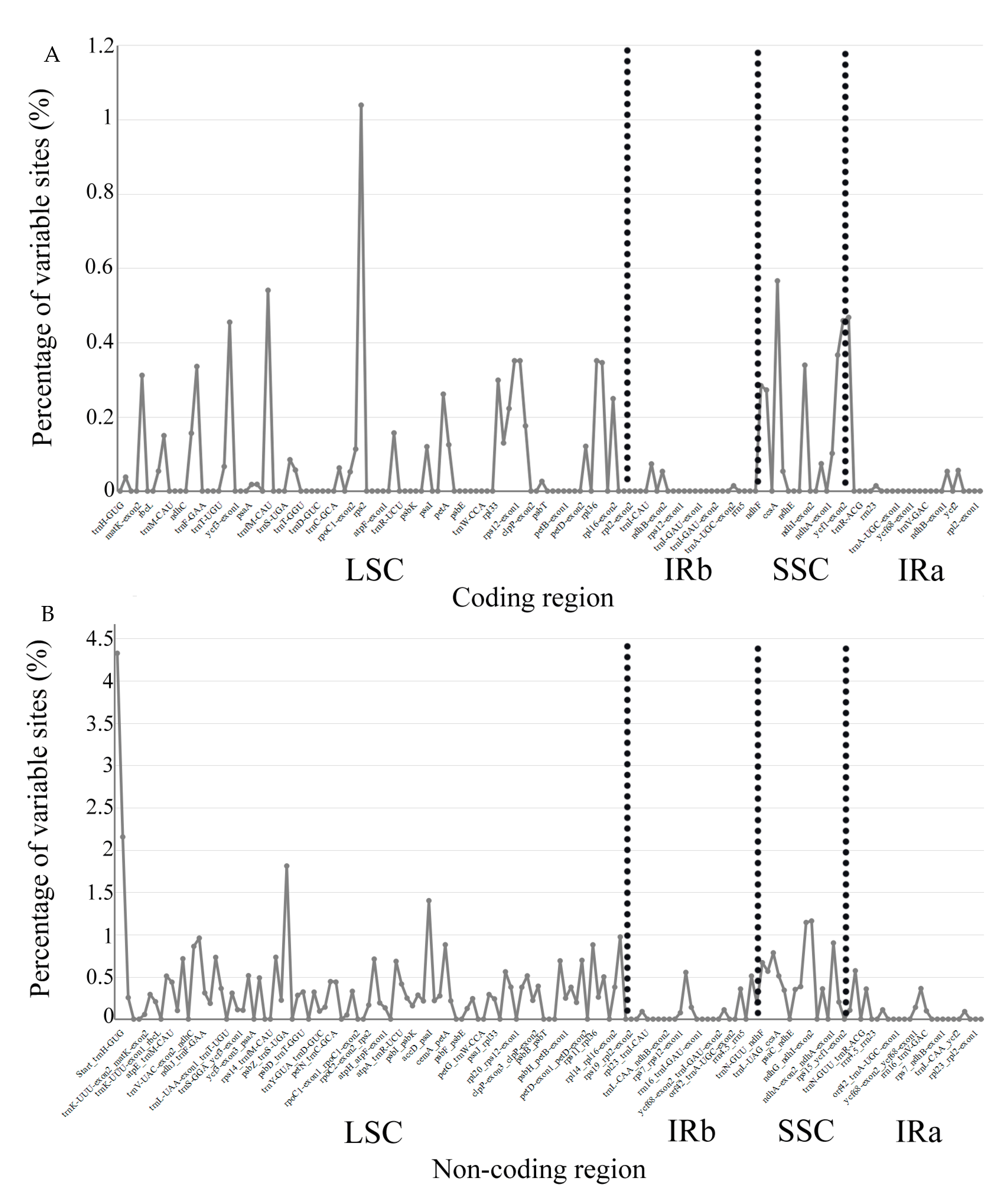
2.4. Sequence Variation of Chloroplast Genome
2.5. Selection on Functional Genes
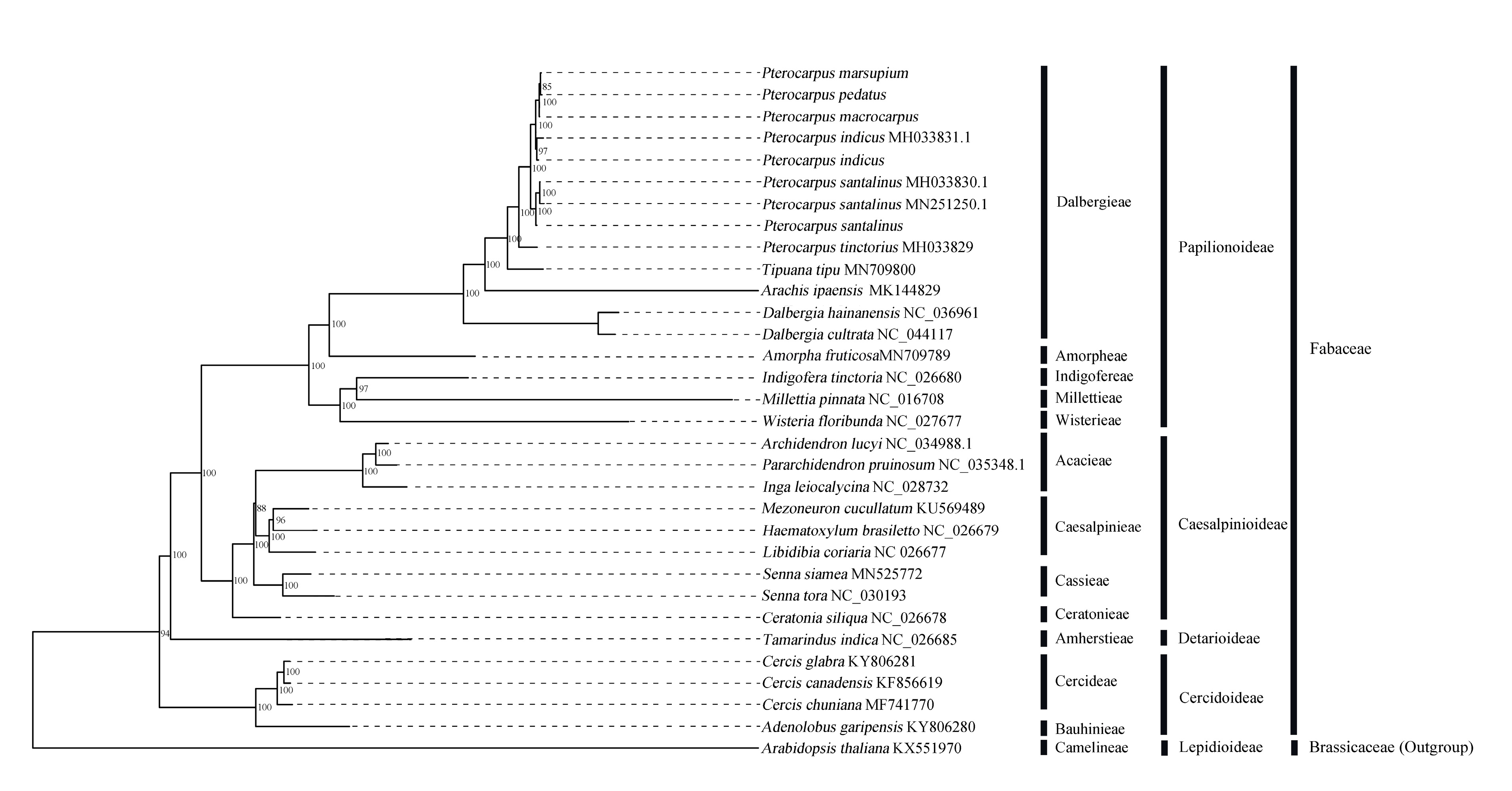
2.6. Phylogenetic Analysis Based on Chloroplast Genome
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. DNA Extraction and Sequencing
4.3. Sequence Assembly of cp Genomes
4.4. Gene Annotation
4.5. Identification of Repeat Sequences and Simple Sequence Repeats (SSR)
4.6. Polymorphism Analysis, Comparison of Genome Structure, and IR Region Contraction and Expansion Analysis of M-S-I-P-M2 cp Genomes
4.7. Gene Selective Pressure Analysis
4.8. Phylogenetic Analysis of Chloroplast Genomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, F.S.P. Pterocarpus Indicus-the Majestic N-fixing Tree; NFT Highlights; Nitrogen Fixing Tree Association (NFTA): Waimanalo, HI, USA, 1992; pp. 92–94. [Google Scholar]
- Jansen, P.C.M.; Westphal, E.; Sosef, M.S.M.; Soerianegara, I.; Lemmens, R. Plant Resources of South-East Asia 5-1. Timber Trees: Major Commercial Timbers; Bogor: Wageningen, The Netherlands, 1993; pp. 102–106. [Google Scholar]
- Chen, Q.D.; Li, X.M.; Zeng, J.; Li, C.W. Introduction status and perspectives of Pterocarpus in China. Sci. Technol. 2004, 2, 38–41. [Google Scholar]
- Saslis-Lagoudakis, C.H.; Klitgaard, B.B.; Forest, F.; Francis, L.; Savolainen, V.; Williamson, E.M.; Hawkins, J.A. The use of phylogeny to interpret cross-cultural patterns in plant use and guide medicinal plant discovery: An example from Pterocarpus (Leguminosae). PLoS ONE 2011, 6, e22275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waeber, P.O.; Schuurman, D.; Ramamonjisoa, B.; Langrand, M.; Barber, C.V.; Innes, J.L.; Lowry II, P.P.; Wilmé, L. Uplisting of Malagasy precious woods critical for their survival. Biol. Conserv. 2019, 235, 89–92. [Google Scholar] [CrossRef]
- Dumenu, W.K. Assessing the impact of felling/export ban and CITES designation on exploitation of African rosewood (Pterocarpus erinaceus). Biol. Conserv. 2019, 236, 124–133. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.; Briscoe, D. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002; p. 617. [Google Scholar]
- IUCN. IUCN red list of threatened species. Version 2017, 2017, 3. [Google Scholar]
- He, T.; Jiao, L.; Wiedenhoeft, A.C.; Yin, Y. Machine learning approaches outperform distance-and tree-based methods for DNA barcoding of Pterocarpus wood. Planta 2019, 249, 1617–1625. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, G.J.; Liu, B.; He, T.; Guo, J.; Jiang, X.; Yin, Y. Wood discrimination analyses of Pterocarpus tinctorius and endangered Pterocarpus santalinus using DART-FTICR-MS coupled with multivariate statistics. IAWA J. 2019, 40, 58–74. [Google Scholar] [CrossRef] [Green Version]
- Yadav, V.K.; Mishra, A. In vitro & in silico study of hypoglycemic potential of Pterocarpus marsupium heartwood extract. Nat. Prod. Res. 2019, 33, 3298–3302. [Google Scholar]
- Karthick, M.; Parthiban, K. Chemical characterization of Pterocarpus santalinus wood using GC-MS. J. Pharmacogn. Phytochem. 2019, 8, 380–382. [Google Scholar]
- Tip-pyang, S.; Aree, T.; Sichaem, J. Pterocarpans and Isoflavones from the Heartwood of Pterocarpus indicus. Chem. Nat. Compd. 2019, 55, 121–123. [Google Scholar] [CrossRef]
- Thanuja, P.; Nadukeri, S.; Kolakar, S.S. Effect of pre sowing seed treatments on seed germination and seedling growth in Rakta Chandana (Pterocarpus santalinus L.): An Endangered medicinal plant. J. For. Environ. Sci. 2019, 7, 1577–1580. [Google Scholar]
- Musa, F.; Jayeoba, F.; Olayiwola, V.; Adekunle, E.; Abiodun, F. Residual effect of organic manure (biochar and cowdung) on the growth of Pterocarpus osun. Curr. J. Appl. Sci. Technol. 2019, 37, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhang, S.; Li, C. Pathogen isolation and identification of a serious leaf disease of red sandalwood (Pterocarpus santalinus) seedlings. Agric. Biotechnol. 2017, 6, 45–48. [Google Scholar]
- Degen, B.; Fladung, M. Use of DNA-markers for tracing illegal logging. In Proceedings of the International Workshop “Fingerprinting Methods for the Identification of Timber Origins”, Bonn, Germany, 8–9 October 2007; pp. 8–9. [Google Scholar]
- Gray, M.W. The evolutionary origins of organelles. Trends Genet. 1989, 5, 294–299. [Google Scholar] [CrossRef]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.R.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.H.; Chan, M.T.; Liao, D.C.; Hsu, C.T.; Lee, Y.W.; Daniell, H.; Duvall, M.R.; Lin, C.S. Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 2010, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Lu, R.S.; Xu, W.Q.; Ohi-Toma, T.; Cai, M.Q.; Qiu, Y.X.; Cameron, K.M.; Fu, C.X. Comparative genomics and phylogenomics of East Asian tulips (Amana, Liliaceae). Front. Plant Sci. 2017, 8, 451. [Google Scholar] [CrossRef] [Green Version]
- Bi, Y.; Zhang, M.F.; Xue, J.; Dong, R.; Du, Y.P.; Zhang, X.H. Chloroplast genomic resources for phylogeny and DNA barcoding: A case study on Fritillaria. Front. Plant Sci. 2018, 8, 1184. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2017, 7, 2054. [Google Scholar] [CrossRef]
- Niu, Y.T.; Florian, J.; Russell, L. Combining complete chloroplast genome sequences with target loci data and morphology to resolve species limits in Triplostegia (Caprifoliaceae). Mol. Phylogenet. Evol. 2018, 129, 15–26. [Google Scholar] [CrossRef]
- Pinard, D.; Myburg, A.A.; Mizrachi, E. The plastid and mitochondrial genomes of Eucalyptus grandis. BMC Genom. 2019, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Coyne, J.A.; Orr, H.A. Speciation; Sinauer Associates: Sunderland, MA, USA, 2004; Volume 276, p. 281. [Google Scholar]
- Mallet, J. Hybrid speciation. Nature 2007, 446, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Borges, L.; Bruneau, A.; Cardoso, D.; Crisp, M.; Delgado-Salinas, A.; Doyle, J.; Egan, A.; Herendeen, P.; Hughes, C.; Kenicer, G. Towards a new classification system for legumes: Progress report from the 6th International Legume Conference. S. Afr. J. Bot. 2013, 89, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Wojciechowski, M.F.; Lavin, M.; Sanderson, M.J. A phylogeny of legumes (Leguminosae) based on analysis of the plastid matK gene resolves many well-supported subclades within the family. Am. J. Bot. 2004, 91, 1846–1862. [Google Scholar] [CrossRef]
- Klitgård, B.B.; Forest, F.; Booth, T.J.; Saslis-Lagoudakis, C.H. A detailed investigation of the Pterocarpus clade (Leguminosae: Dalbergieae): Etaballia with radially symmetrical flowers is nested within the papilionoid-flowered Pterocarpus. S. Afr. J. Bot. 2013, 89, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Jiao, L.; Lu, Y.; He, T.; Li, J.; Yin, Y. A strategy for developing high-resolution DNA barcodes for species discrimination of wood specimens using the complete chloroplast genome of three Pterocarpus species. Planta 2019, 250, 95–104. [Google Scholar] [CrossRef]
- Huang, J.; Chen, R.; Li, X. Comparative analysis of the complete chloroplast genome of four known Ziziphus species. Genes 2017, 8, 340. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.M.; Liu, H.M.; Hu, J.J.; Liang, Y.Q.; Liang, J.J.; Wu, T.N. Five complete chloroplast genome sequences from Diospyros: Genome organization and comparative analysis. PLoS ONE 2016, 11, e0159566. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.P.; Meng, Y.; Sun, H.; Nie, Z.L. Molecular phylogeny of Gueldenstaedtia and Tibetia (Fabaceae) and their biogeographic differentiation within Eastern Asia. PLoS ONE 2006, 11, e0162982. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, L.; Yan, T.L.; Liu, Q. Complete chloroplast genome sequences of Praxelis (Eupatorium catarium Veldkamp), an important invasive species. Gene 2014, 549, 58–69. [Google Scholar] [CrossRef]
- Curci, P.L.; De Paola, D.; Danzi, D.; Vendramin, G.G.; Sonnante, G. Complete chloroplast genome of the multifunctional crop globe artichoke and comparison with other Asteraceae. PLoS ONE 2015, 10, e0120589. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Messing, J. High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, W.L.; Cai, Y.; Wu, W.; Zhou, R. The complete chloroplast genome sequence of Melastoma candidum (Melastomataceae). Mitochondrial DNA Part B 2017, 2, 242–243. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Tembrock, L.R.; Zheng, S.; Wu, Z. The complete chloroplast genome of Catha edulis: A comparative analysis of genome features with related species. Int. J. Mol. Sci. 2018, 19, 525. [Google Scholar] [CrossRef] [Green Version]
- Yi, D.K.; Kim, K.J.; Zhang, J.W. Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 2012, 7, e35872. [Google Scholar] [CrossRef] [Green Version]
- Mariotti, R.; Cultrera, N.G.; Díez, C.M.; Baldoni, L.; Rubini, A. Identification of new polymorphic regions and differentiation of cultivated olives (Olea europaea L.) through plastome sequence comparison. BMC Plant Biol. 2010, 10, 211. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Flegel, W.A. Codon usage in vertebrates is associated with a low risk of acquiring nonsense mutations. J. Transl. Med. 2011, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.L.; Ma, L.; Gong, P.; Liu, X.Q.; Wang, Z.; Zhao, GQ. Development and application of EST–SSRs markers for analysis of genetic diversity in erect milkvetch (Astragalus adsurgens Pall.). Mol. Biol. Rep. 2019, 46, 1323–1326. [Google Scholar] [CrossRef]
- Pauwels, M.; Vekemans, X.; Godé, C.; Frérot, H.; Castric, V.; Saumitou-Laprade, P. Nuclear and chloroplast DNA phylogeography reveals vicariance among European populations of the model species for the study of metal tolerance, Arabidopsis halleri (Brassicaceae). New Phytol. 2012, 193, 916–928. [Google Scholar] [CrossRef]
- Zhao, K.K.; Landrein, S.; Barrett, R.; Sakaguchi, S.; Maki, M.; Mu, W.; Yang, T.; Zhu, Z.; Liu, H.; Wang, H.F. Phylogeographic analysis and genetic structure of an endemic Sino-Japanese disjunctive genus Diabelia (Caprifoliaceae). Front. Plant Sci. 2019, 10, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonah, H.; Deshmukh, R.K.; Sharma, A.; Singh, V.P.; Gupta, D.K.; Gacche, R.N.; Rana, J.C.; Singh, N.K.; Sharma, T.R. Genome-wide distribution and organization of microsatellites in Plants: An insight into marker development in Brachypodium. PLoS ONE 2011, 6, e21298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.L.; Xu, L.A.; Huan, M.R.; Wang, Z.R. Genetic diversity of microsatellites (SSRs) of natural populations of Quercus variabilis. Hereditas (Beijing) 2004, 26, 683–688. [Google Scholar]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Schlotterer, C.; Tautz, D. Slippage synthesis of simple sequence DNA. Nucleic Acids Res. 1992, 20, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.M. Microsatellites and other simple sequences: Genomic context and mutational mechanisms. In Microsatellites: Evolution and Applications; Goldstein, D., Schlotterer, C., Eds.; Oxford University Press: New York, NY, USA, 1999; pp. 1–9. [Google Scholar]
- Wang, L.Y.; Liu, B.Y.; Jiang, Y.H.; Duan, Y.S.; Cheng, H.; Zhou, J.; Tang, Y.C. Phylogenetic analysis of interspecies in section Thea through SSR markers. J. Tea Sci. 2009, 29, 341–346. [Google Scholar]
- Shi, J.X.; Qiao, Y.; Ma, Y.Q.; Ji, W.Q.; He, P.R.; Weng, Y.J. Analysis on genetic evolution relation of A, B genomes between Triticum aestivum and T. dicoccoides by SSR. Acta Bot. Boreali Occident. Sin. 2003, 23, 933–937. [Google Scholar]
- Deng, S.Y.; Wen, Q.; Li, K.Q.; Ye, J.S.; Zhu, P.L. Screening and application of SSR primers in plants of Clerodendrum L. Chin. Tradit. Herb. Drugs 2014, 63, 3317–3322. [Google Scholar]
- Han, Q.; Wang, H.; Wang, J.M.; Zhao, J.R.; Yang, M.-S. Genertic diversity analysis of salicaceae cultivars by transferablity of SSR primers. Mol. Plant Breed. 2009, 5, 904–911. [Google Scholar]
- Zhong, M.; Cheng, X.Z.; Wang, L.X.; Wang, S.H.; Wang, X. Transferability of mungbean genomic-SSR markers in other Vigna species. Acta Agron. Sin. 2012, 38, 223–230. [Google Scholar] [CrossRef]
- Maréchal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.H.; Lou, W.R.; Chen, Y.Q.; Jia, X.D.; Zhai, M.; Guo, Z.R.; Xuan, J.P. The Chloroplast Genome of Carya illinoinensis: Genome Structure, Adaptive Evolution, and Phylogenetic Analysis. Forests 2020, 11, 207. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, D.; Senalik, D.; Ames, M.; Zhu, H.; Steffan, S.A.; Harbut, R.; Polashock, J.; Vorsa, N.; Gillespie, E.; Kron, K. Complete plastid genome sequence of Vaccinium macrocarpon: Structure, gene content, and rearrangements revealed by next generation sequencing. Tree Genet. Genomes 2013, 9, 489–498. [Google Scholar] [CrossRef]
- Kuang, D.Y.; Wu, H.; Wang, Y.L.; Gao, L.M.; Zhang, S.Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): Comparative analyses and identification of mutational hotspots. Genomics 2020, 112, 581–591. [Google Scholar]
- Clegg, M.T.; Gaut, B.S.; Learn, G.H.; Morton, B.R. Rates and patterns of chloroplast DNA evolution. Proc. Natl. Acad. Sci. USA 1994, 91, 6795–6801. [Google Scholar] [CrossRef] [Green Version]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.-Y.; Gao, L.-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.F.; Wu, M.L.; Liao, B.S.; Liu, Z.X.; Bai, R.; Xiao, S.M.; Li, X.W.; Zhang, B.L.; Xu, J.; Chen, S.L. Complete chloroplast genome sequence and phylogenetic analysis of the medicinal plant Artemisia annua. Molecules 2017, 22, 1330. [Google Scholar] [CrossRef]
- Dong, W.P.; Liu, H.; Xu, C.; Zuo, Y.J.; Chen, Z.J.; Zhou, S.L. A chloroplast genomic strategy for designing taxon specific DNA mini-barcodes: A case study on ginsengs. BMC Genet. 2014, 15, 138. [Google Scholar] [CrossRef] [Green Version]
- Chat, J.; Decroocq, S.; Decroocq, V.; Petit, R. A case of chloroplast heteroplasmy in kiwifruit (Actinidia deliciosa) that is not transmitted during sexual reproduction. J. Hered. 2002, 93, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.Y.; Zheng, X.Y.; Teng, Y.W. Characterization and phylogenetic utility of non-coding chloroplast regions trnL-trnF and accD-psaI in Pyrus. Acta Hortic. Sin. 2011, 38, 2261–2272. [Google Scholar]
- Liston, A.; Wheeler, J.A. The phylogenetic position of the genus Astragalus (Fabaceae): Evidence from the chloroplast genes rpoC1 and rpoC2. Biochem. Syst. Ecol. 1994, 22, 377–388. [Google Scholar] [CrossRef]
- Scarcelli, N.; Barnaud, A.; Eiserhardt, W.; Treier, U.A.; Seveno, M.; d’Anfray, A.; Vigouroux, Y.; Pintaud, J.-C. A set of 100 chloroplast DNA primer pairs to study population genetics and phylogeny in monocotyledons. PLoS ONE 2011, 6, e19954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Li, X.; Zhu, X.; Huang, X.; Jin, S. The Complete Plastid Genome of Rhododendron pulchrum and Comparative Genetic Analysis of Ericaceae Species. Forests 2020, 11, 158. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.X.; Li, R.; Worth, J.R.; Li, X.; Li, P.; Cameron, K.M.; Fu, C.X. The complete chloroplast genome of Chinese bayberry (Morella rubra, Myricaceae): Implications for understanding the evolution of Fagales. Front. Plant Sci. 2017, 8, 968. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Jansen, R.K. Chloroplast genomes of plants. In Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; Henry, R.J., Ed.; CABI Publishing: Wallingford, UK, 2005; pp. 45–68. [Google Scholar]
- Muse, S.V.; Gaut, B.S. Comparing patterns of nucleotide substitution rates among chloroplast loci using the relative ratio test. Genetics 1997, 146, 393–399. [Google Scholar]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [Green Version]
- Timmis, J.N.; Ayliffe, M.A.; Huang, C.-Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef]
- Erixon, P.; Oxelman, B. Whole-gene positive selection, elevated synonymous substitution rates, duplication, and indel evolution of the chloroplast clpP1 gene. PLoS ONE 2008, 3, e1386. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Kong, J.J.; Qiu, J.L.; Zhu, H.S.; Peng, Y.C.; Jiang, H.Y. High level of microsynteny and purifying selection affect the evolution of WRKY family in Gramineae. Dev. Genes Evol. 2016, 226, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.K.; Purugganan, M.D. Evolutionary and ecological genomics of Arabidopsis. Plant Physiol. 2005, 138, 578–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, M.; Pennington, R.T.; Klitgaard, B.B.; Sprent, J.I.; de Lima, H.C.; Gasson, P.E. The dalbergioid legumes (Fabaceae): Delimitation of a pantropical monophyletic clade. Am. J. Bot. 2001, 88, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Kajita, T.; Ohashi, H.; Tateishi, Y.; Bailey, C.D.; Doyle, J.J. rbcL and legume phylogeny, with particular reference to Phaseoleae, Millettieae, and allies. Syst. Bot. 2001, 26, 515–536. [Google Scholar]
- Compton, J.A.; Schrire, B.D.; Könyves, K.; Forest, F.; Malakasi, P.; Mattapha, S.; Sirichamorn, Y. The Callerya Group redefined and Tribe Wisterieae (Fabaceae) emended based on morphology and data from nuclear and chloroplast DNA sequences. PhytoKeys 2019, 125, 1. [Google Scholar] [CrossRef]
- McPherson, H.; Van der Merwe, M.; Delaney, S.K.; Edwards, M.A.; Henry, R.J.; McIntosh, E.; Rymer, P.D.; Milner, M.L.; Siow, J.; Rossetto, M. Capturing chloroplast variation for molecular ecology studies: A simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 2013, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Borgstrom, E.; Lundin, S.; Lundeberg, J. Large scale library generation for high throughput sequencing. PLoS ONE 2011, 6, e19119. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Y.P.; Peluso, P.; Shi, J.H.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524. [Google Scholar] [CrossRef]
- Winiarski, M.; Kuśmirek, W.; Nowak, R.M. De Novo DNA assembler for third generation sequencers’ reads based on BLASR algorithm. In Photonics Applications in Astronomy, Communications, Industry, and High-Energy Physics Experiments 2018; International Society for Optics and Photonics: Bellingham, WA, USA, 2018; p. 108083C. [Google Scholar]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 18. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kanehisa, M. A database for post-genome analysis. Trends Genet. Tig 1997, 13, 375. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, 354–357. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Magrane, M. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A.; Thiel, T. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.Z.; Kong, H.H.; Zhou, J.; Fritsch, P.W.; Hao, G.; Gong, W. Complete chloroplast genome of Cercis chuniana (Fabaceae) with structural and genetic comparison to six species in Caesalpinioideae. Int. J. Mol. Sci. 2018, 19, 1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Gateway Computing Environments Workshop; IEEE Computer Society: New Orleans, LA, USA, 2010; pp. 1–8. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Tembrock, L.R.; Ge, S. Are differences in genomic data sets due to true biological variants or errors in genome assembly: An example from two chloroplast genomes. PLoS ONE 2015, 10, e0118019. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Group | Genes |
|---|---|---|
| Photosynthesis | Subunits_of_photosystem_I | psaA-, psaB, psaI, psaJ, psaC |
| Subunits_of_photosystem_II | psbA, psbZ, psbC-, psbD, psbM, psbI, psbK, psbL, psbF, psbE, psbB-, psbT, psbN, psbH | |
| Subunits_of_NADH_dehydrogenase | ndhC, ndhK, ndhJ, ndhB(×2) * +, ndhF, ndhD+, ndhE, ndhG, ndhI *, ndhA *, ndhH- | |
| Subunits_of_cytochrome_b/f_complex | petN, petA, petL, petG, petB *, petD * | |
| Subunits_of_ATP_synthase | atpB-, atpE, atpI, atpH, atpF *, atpA | |
| Large_subunit_of_Rubisco | rbcL | |
| Self-replication | Large_subunits_of_ribosome | rpl33, rpl20, rpl36-, rpl14, rpl16 *, rpl2(×2) *, rpl32+, rpl23(×2) |
| Small_subunits_of_ribosome | rps4+, rps14, rps2, rps18, rps11, rps8, rps3, rps19, rps12 *- (×2,part), rps15, rps7(×2) | |
| DNA-dependent_RNA_polymerase | rpoB, rpoC1 *, rpoC2 *, rpoA | |
| Ribosomal_RNAs | rrn5, rrn4.5, rrn23, rrn16 | |
| Transfer_RNAs | trnH-GUG, trnK-UUU *, trnM-CAU, trnV-UAC *, trnF-GAA, trnL-UAA *, trnT-UGU, trnS-GGA, trnfM-CAU, trnG-UCC, trnS-UGA, trnT-GGU, trnE-UUC, trnY-GUA, trnD-GUC, trnC-GCA, trnR-UCU, trnS-GCU, trnQ-UUG, trnW-CCA, trnP-UGG, trnI-CAU, trnL-CAA, trnV-GAC, trnI-GAU *, trnA-UGC *, trnR-ACG, trnN-GUU, trnL-UAG | |
| Other genes | Maturase | matK * |
| Protease | clpP **- | |
| Envelope_membrane_protein | cemA | |
| Acetyl-CoA_carboxylase | accD+ | |
| C-type_cytochrome_synthesis_gene | ccsA | |
| Genes of unknown function | Proteins_of_unknown_function | ycf3 **, ycf4, ycf68(×2) *, ycf1, orf42(×2), ycf2(×2) + |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Z.; Wu, Z.; Zhao, K.; Yang, Z.; Zhang, N.; Guo, J.; Tembrock, L.R.; Xu, D. Comparative Analyses of Five Complete Chloroplast Genomes from the Genus Pterocarpus (Fabacaeae). Int. J. Mol. Sci. 2020, 21, 3758. https://doi.org/10.3390/ijms21113758
Hong Z, Wu Z, Zhao K, Yang Z, Zhang N, Guo J, Tembrock LR, Xu D. Comparative Analyses of Five Complete Chloroplast Genomes from the Genus Pterocarpus (Fabacaeae). International Journal of Molecular Sciences. 2020; 21(11):3758. https://doi.org/10.3390/ijms21113758
Chicago/Turabian StyleHong, Zhou, Zhiqiang Wu, Kunkun Zhao, Zengjiang Yang, Ningnan Zhang, Junyu Guo, Luke R. Tembrock, and Daping Xu. 2020. "Comparative Analyses of Five Complete Chloroplast Genomes from the Genus Pterocarpus (Fabacaeae)" International Journal of Molecular Sciences 21, no. 11: 3758. https://doi.org/10.3390/ijms21113758