Characterization of Nuclear and Mitochondrial Genomes of Two Tobacco Endophytic Fungi Leptosphaerulina chartarum and Curvularia trifolii and Their Contributions to Phylogenetic Implications in the Pleosporales


Abstract
:1. Introduction
2. Results
2.1. Fungal Isolation and Identification
2.2. Nuclear Genome Features of L. chartarum and C. trifolii
2.3. General Features of the Newly Sequenced Mitochondrial Genomes
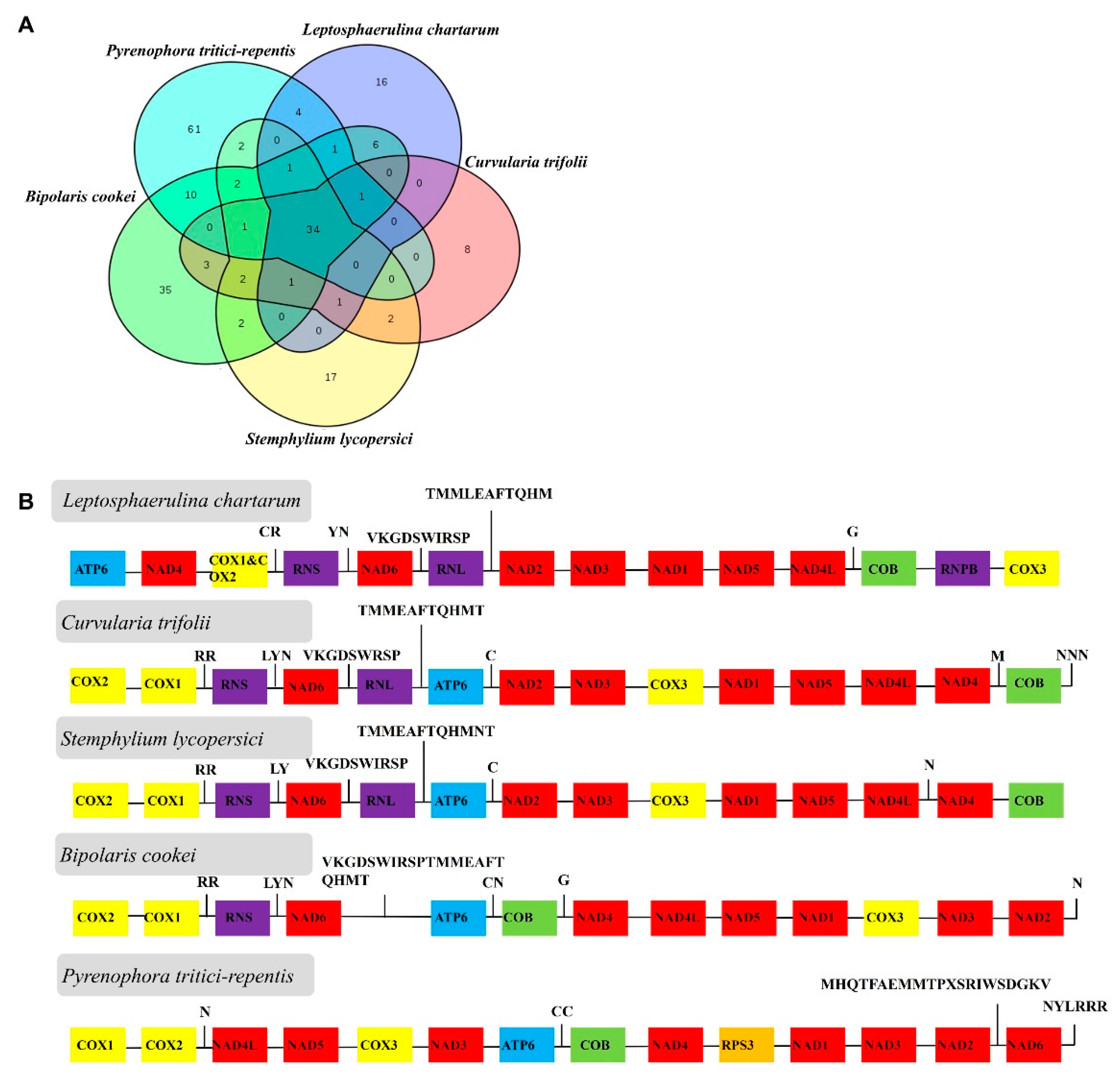
2.4. Genome Comparison
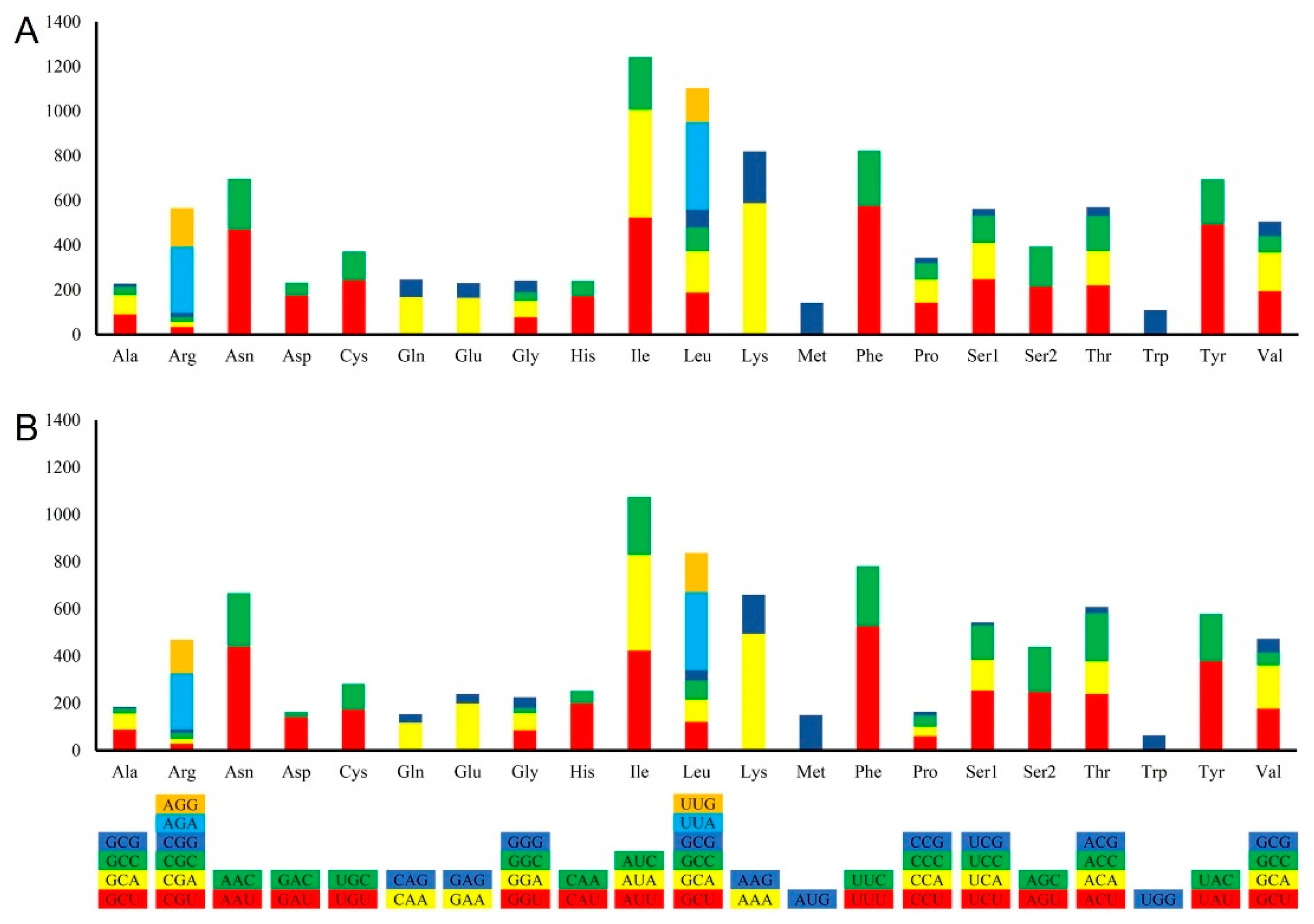
2.5. Evolutionary Rates of Nuclear and Mitochondrial Genes
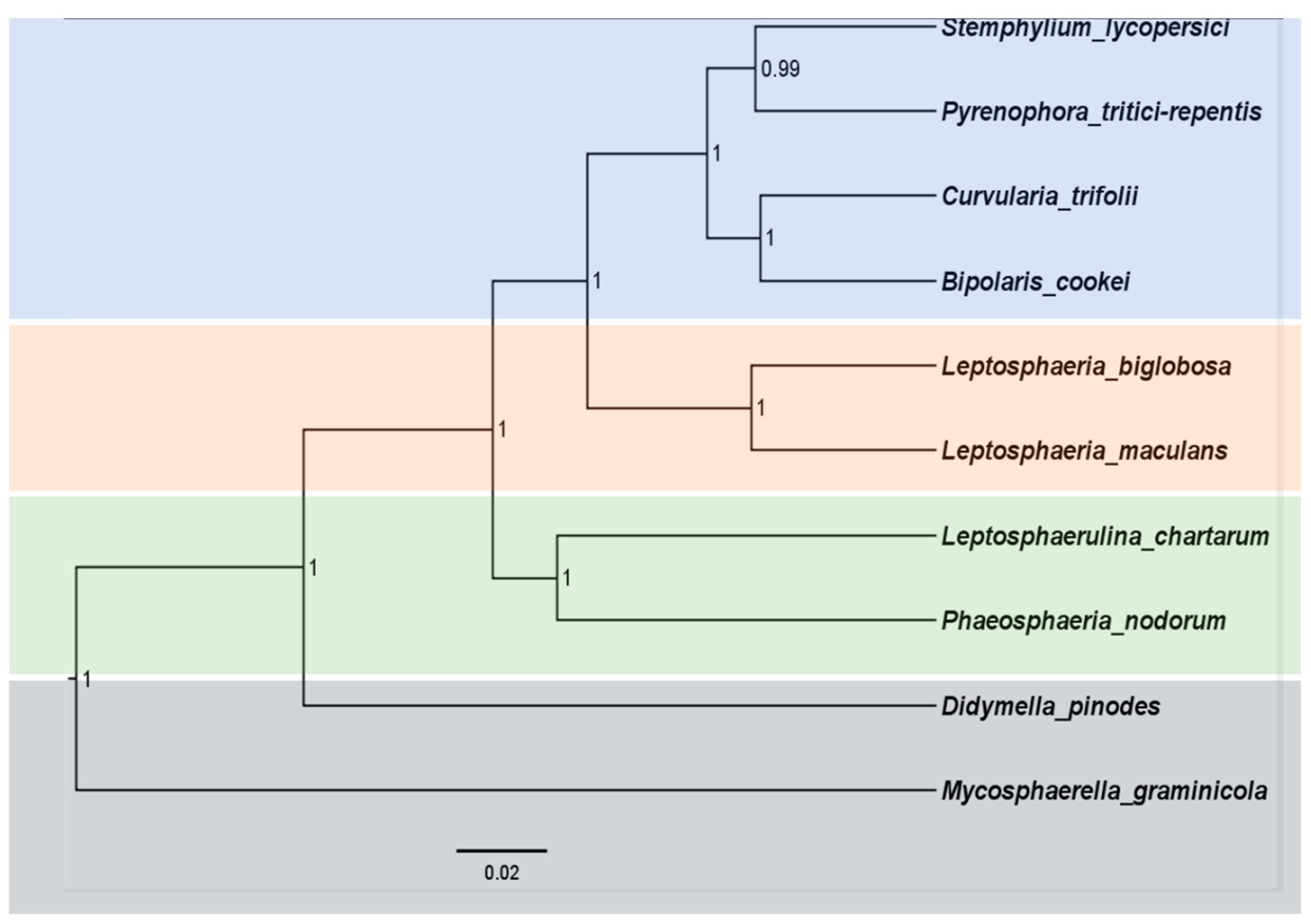
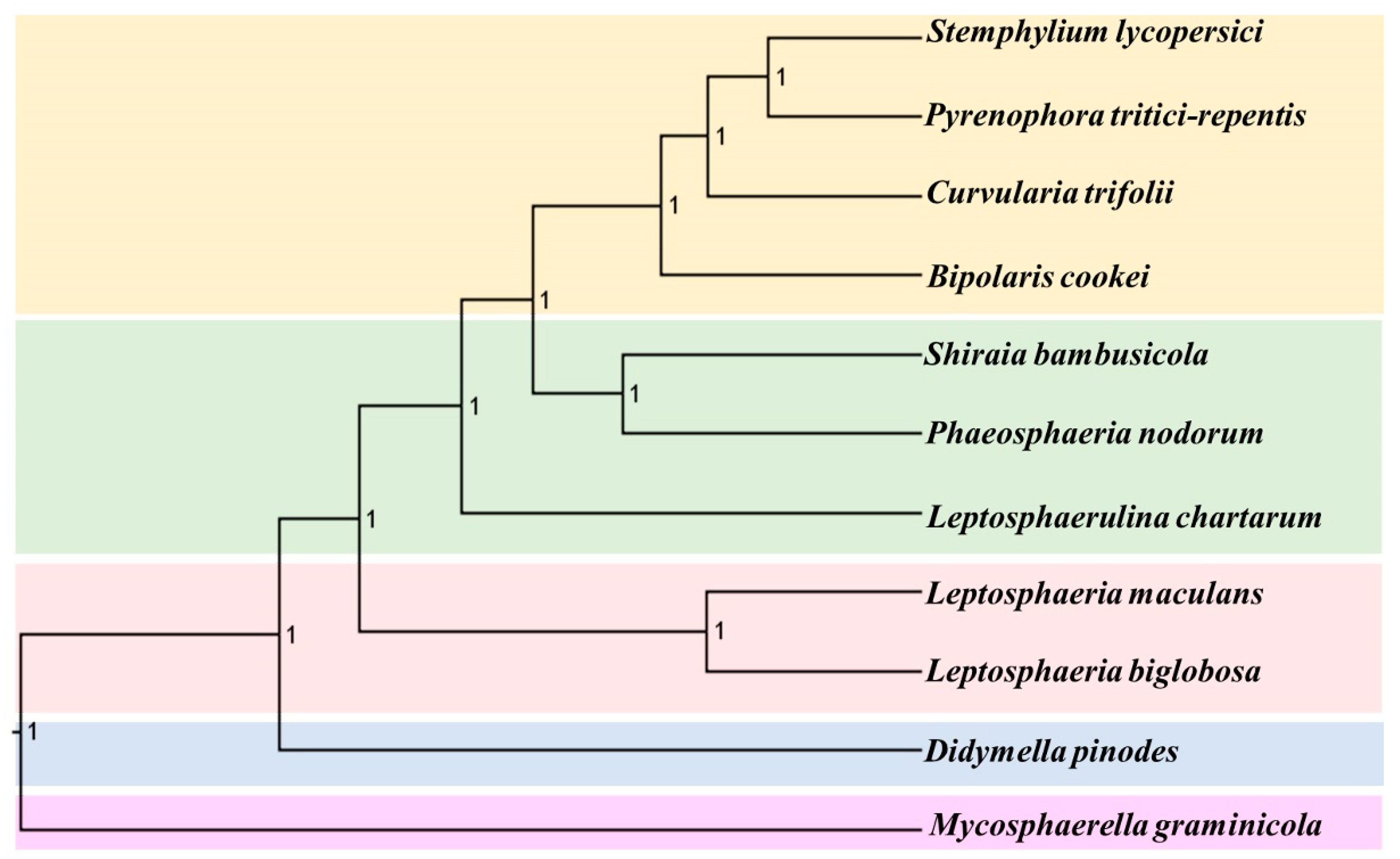
2.6. Phylogenetic Relationship
3. Discussion
4. Materials and Methods
4.1. Materials, Isolation, and Culture
4.2. DNA Extraction
4.3. Nuclear and Mitochondrial Genome Assembly
4.4. Genome Prediction, Annotation and Analysis
4.5. Evolutionary Rates of the Nuclear Genes and Mitochondrial Genes
4.6. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Spurr, H.W., Jr.; Welty, R.E. Characterization of endophytic fungi in healthy leaves of Nicotiana spp. Phytopathology 1975, 65, 417–422. [Google Scholar] [CrossRef]
- Jin, H.Q.; Liu, H.B.; Xie, Y.Y.; Zhang, Y.G.; Xu, Q.Q.; Mao, L.J.; Li, X.J.; Chen, J.; Lin, F.C.; Zhang, C.L. Effect of the dark septate endophytic fungus Acrocalymma vagum on heavy metal content in tobacco leaves. Symbiosis 2018, 74, 89–95. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, Y.C. Tobacco Growth Promotion by the Entomopathogenic Fungus, Isaria javanica pf185. Mycobiology 2019, 47, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanasinghe, D.N.; Jeewon, R.; Jones, E.G.; Boonmee, S.; Kaewchai, S.; Manawasinghe, I.S.; Lumyong, S.; Hyde, K.D. Novel palmicolous taxa within Pleosporales: Multigene phylogeny and taxonomic circumscription. Mycol. Prog. 2018, 17, 571–590. [Google Scholar] [CrossRef]
- Yuan, X.L.; Cao, M.; Liu, X.M.; Du, Y.M.; Shen, G.M.; Zhang, Z.F.; Li, J.H.; Zhang, P. Composition and genetic diversity of the Nicotiana tabacum microbiome in different topographic areas and growth periods. Int. J. Mol. Sci. 2018, 19, 3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodsueb, R.; Dhanasekaran, V.; Aptroot, A.; Lumyong, S.; McKenzie, E.H.; Hyde, R.; Jeewon, K.D. The family Pleosporaceae: Intergeneric relationships and phylogenetic perspectives based on sequence analyses of partial 28S rDNA. Mycologia 2006, 98, 571–583. [Google Scholar] [CrossRef]
- Eken, C.; Jochum, C.C.; Yuen, G.Y. First report of leaf spot of smooth bromegrass caused by Pithomyces chartarum in Nebraska. Plant Dis. 2006, 90, 108. [Google Scholar] [CrossRef]
- Vu, A.L.; Gwinn, K.D.; Ownley, B.H. First Report of Leaf Spot on Switchgrass Caused by Pithomyces chartarum in the United States. Plant Dis. 2013, 97, 1655. [Google Scholar] [CrossRef]
- Verma, R.K.; Sharma, A.K.; Gupta, B.D. Surface plasmon resonance based tapered fiber optic sensor with different taper profiles. Opt. Commun. 2008, 281, 1486–1491. [Google Scholar] [CrossRef]
- Ahonsi, M.O.; Ames, K.A.; Gray, M.E.; Bradley, C.A. Biomass reducing potential and prospective fungicide control of a new leaf blight of Miscanthus× giganteus caused by Leptosphaerulina chartarum. Bioenerg. Res. 2013, 6, 737–745. [Google Scholar] [CrossRef] [Green Version]
- Perelló, A.; Aulicino, M.; Stenglein, S.A.; Labuda, R.; Moreno, M.V. Pseudopithomyces chartarum associated with wheat seeds in Argentina, pathogenicity and evaluation of toxigenic ability. Eur. J. Plant Pathol. 2017, 148, 491–496. [Google Scholar] [CrossRef]
- Zhu, Y.; Hassan, Y.I.; Watts, C.; Zhou, T. Innovative technologies for the mitigation of mycotoxins in animal feed and ingredients—A review of recent patents. Anim. Feed Sci. Technol. 2016, 216, 19–29. [Google Scholar] [CrossRef]
- Matthews, Z.M.; Edwards, P.J.; Kahnt, A.; Collett, M.G.; Marshall, J.C.; Partridge, A.C.; Harrison, S.J.; Fraser, K.; Cao, M.; Derrick, P.J. Serum metabolomics using ultra performance liquid chromatography coupled to mass spectrometry in lactating dairy cows following a single dose of sporidesmin. Metabolomics 2018, 14, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.; Recer, G.; Hwang, S.; Lin, S. Association between indoor mold and asthma among children in Buffalo, New York. Indoor Air 2011, 21, 156–164. [Google Scholar] [CrossRef]
- Dannemiller, K.C.; Gent, J.F.; Leaderer, B.P.; Peccia, J. Influence of housing characteristics on bacterial and fungal communities in homes of asthmatic children. Indoor Air 2016, 26, 179–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, C.H.; Koo, J.H.; Kim, J.H.; Yoon, J.H.; Lee, J.H.; Shim, K.Y.; Kwak, Y.S.; Chang, S.W. First report of Curvularia leaf blight caused by Curvularia trifolii on Creeping bentgrass in Korea. Weed Turfgrass Sci. 2016, 5, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, J.; Cheng, J.; Zheng, C.; Li, W.; Wang, Y.C. Leaf Spot of Tobacco (Nicotiana tabacum) Caused by Curvularia trifolii in Guizhou Province of China. Plant Dis. 2017, 101, 1549. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Ran, S.F.; Bhat, J.; Hyde, K.D.; Wang, Y.; Zhao, D.G. Curvularia microspora sp. nov. associated with leaf diseases of Hippeastrum striatum in China. MycoKeys 2018, 29, 49. [Google Scholar] [CrossRef] [Green Version]
- Couttolenc, A.; Espinoza, C.; Fernández, J.J.; Norte, M.; Plata, G.B.; Padrón, J.M.; Shnyreva, A.; Trigos, Á. Antiproliferative effect of extract from endophytic fungus Curvularia trifolii isolated from the “Veracruz Reef System” in Mexico. Pharm. Biol. 2016, 54, 1392–1397. [Google Scholar] [CrossRef] [Green Version]
- Samanthi, K.A.U.; Wickramaarachchi, S.; Wijeratne, E.M.K.; Paranagama, P.A. Two new bioactive polyketides from Curvularia trifolii, an endolichenic fungus isolated from Usnea sp.; in Sri Lanka. J. Natl. Sci. Found. Sri. Lanka 2015, 43, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Ahonsi, M.; Agindotan, B.; Williams, D.; Arundale, R.; Gray, M.; Voigt, T.; Bradley, C.A. First report of Pithomyces chartarum causing a leaf blight of Miscanthus× giganteus in Kentucky. Plant Dis. 2010, 94, 480. [Google Scholar] [CrossRef] [PubMed]
- Jayasiri, S.C.; Gareth, J.E.B.; Kang, J.C.; Promputtha, I.; Bahkali, A.H.; Sar, K.D.H.; Zhang, Y.; Wang, H.; Fournier, J.; Crous, P.; et al. A new species of genus Anteaglonium (Anteagloniaceae, Pleosporales) with its asexual morph. Phytotaxa 2016, 263, 233–244. [Google Scholar] [CrossRef]
- Nuankaew, S.; Suetrong, S.; Wutikhun, T.; Wutikhun, T.; Pinruan, U. Hermatomyces trangensis sp. nov.; a new dematiaceous hyphomycete (Hermatomycetaceae, Pleosporales) on sugar palm in Thailand. Phytotaxa 2019, 391, 277–288. [Google Scholar] [CrossRef]
- Liu, Y.; Johnson, M.G.; Cox, C.J.; Medina, R.; Devos, N.; Vanderpoorten, A.; Hedenäs, L.; Bell, N.E.; Shevock, J.R.; Aguero, B.; et al. Resolution of the ordinal phylogeny of mosses using targeted exons from organellar and nuclear genomes. Nat. Commun. 2019, 10, 1–11. [Google Scholar]
- Williams, T.A.; Cox, C.J.; Foster, P.G.; Szöllősi, G.J.; Embley, T.M. Phylogenomics provides robust support for a two-domains tree of life. Nat. Ecol. Evol. 2020, 4, 138–147. [Google Scholar] [CrossRef]
- Schober, A.F.; Río Bártulos, C.; Bischoff, A.; Lepetit, B.; Gruber, A.; Kroth, P.G. Organelle studies and proteome analyses of mitochondria and plastids fractions from the diatom Thalassiosira pseudonana. Plant Cell Physiol. 2019, 60, 1811–1828. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Chaturvedi, P.; Kulkarni, M.G.; Van Staden, J. A critical review on exploiting the pharmaceutical potential of plant endophytic fungi. Biotechnol. Adv. 2019, 39, 107462. [Google Scholar] [CrossRef]
- Dastogeer, K.M.G.; Li, H.; Sivasithamparam, K.; Jones, M.G.K.; Wylie, S.J. Host specificity of endophytic mycobiota of wild Nicotiana plants from arid regions of northern Australia. Microb. Ecol. 2018, 75, 74–87. [Google Scholar] [CrossRef]
- Wu, Q.; Li, Y.; Li, Y.; Gao, S.; Wang, M.; Zhang, T.; Chen, J. Identification of a novel fungus, Leptosphaerulina chartarum SJTU59 and characterization of its xylanolytic enzymes. PLoS ONE 2013, 8, e73729. [Google Scholar] [CrossRef]
- Zhang, P.; Li, J.; Lang, J.; Jia, C.; Niaz, S.I.; Chen, S.; Liu, L. Two new sesquiterpenes derivatives from marine fungus Leptosphaerulina Chartarum sp. 3608. Nat. Prod. Res. 2018, 32, 2297–2303. [Google Scholar] [CrossRef]
- Torriani, S.F.; Goodwin, S.B.; Kema, G.H.; Pangilinan, J.L.; McDonald, B.A. Biology, Intraspecific comparison and annotation of two complete mitochondrial genome sequences from the plant pathogenic fungus Mycosphaerella graminicola. Fungal. Genet. Biol. 2008, 45, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Haridas, S.; Albert, R.; Binder, M.; Bloem, J.; LaButti, K.; Salamov, A.; Andreopoulos, B.; Baker, S.E.; Barry, K.; Bills, G.; et al. 101 Dothideomycetes genomes: A test case for predicting lifestyles and emergence of pathogens. Stud. Mycol. 2020, 96, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.Y.; Li, T.; Chen, S.; Fan, L.; Gao, J.; Hou, C.L. Characterization and phylogenetic analysis of the mitochondrial genome of Shiraia bambusicola reveals special features in the order of Pleosporales. PLoS ONE 2015, 10, e0116466. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Cao, L. Fungal endophytes and their interactions with plants in phytoremediation: A review. Chemosphere 2017, 168, 1100–1106. [Google Scholar] [CrossRef]
- Ng, K.P.; Yew, S.M.; Chan, C.L.; Soo-Hoo, T.S.; Na, S.L.; Hassan, H.; Ngeow, Y.F.; Hoh, C.C.; Lee, K.W.; Yee, W.Y. Genome sequence of an unclassified pleosporales species isolated from human nasopharyngeal aspirate. Eukaryot. Cell 2012, 11, 828. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Liu, Y.; Tan, Y.; Huang, Y.; Liu, Z.; Jiang, X. Sequencing and functional annotation of the whole genome of Shiraia bambusicola. G3 (Bethesda) 2020, 10, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Ohm, R.A.; Feau, N.; Henrissat, B.; Schoch, C.L.; Horwitz, B.A.; Barry, K.W.; Condon, B.J.; Copeland, A.C.; Dhillon, B.; Glaser, F.; et al. Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog. 2012, 8, e1003037. [Google Scholar] [CrossRef] [Green Version]
- Losada, L.; Pakala, S.B.; Fedorova, N.D.; Joardar, V.; Shabalina, S.A.; Hostetler, J.; Pakala, S.M.; Zafar, N.; Thomas, E.; Rodriguez-Carres, M.; et al. Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 2014, 352, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Koszul, R.; Malpertuy, A.; Frangeul, L.; Bouchier, C.; Wincker, P.; Thierry, A.; Duthoy, S.; Ferris, S.; Hennequin, C.; Dujon, B. The complete mitochondrial genome sequence of the pathogenic yeast Candida (Torulopsis) glabrata. FEBS Lett. 2003, 534, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Oh, J.; Kong, W.S.; Sung, G.H. Complete mitochondrial genome of the entomopathogenic fungus Beauveria pseudobassiana (Ascomycota, Cordycipitaceae). Mitochondrial DNA 2015, 26, 777–778. [Google Scholar] [CrossRef]
- Van de Sande, W.W.J. Phylogenetic analysis of the complete mitochondrial genome of Madurella mycetomatis confirms its taxonomic position within the order Sordariales. PLoS ONE 2012, 7, e38654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, E.R.; Adams, A.N.; Spotten, S.M.; Van Hove, R.A.; Whittington, K.T.; Keepers, K.G.; Pogoda, C.S.; Lendemer, J.C.; Tripp, E.A.; Kane, N.C. The complete mitochondrial genomes of five lichenized fungi in the genus Usnea (Ascomycota: Parmeliaceae). Mitochondrial DNA Part B 2018, 3, 305–308. [Google Scholar] [CrossRef] [Green Version]
- Sandor, S.; Zhang, Y.; Xu, J. Fungal mitochondrial genomes and genetic polymorphisms. Appl. Microbiol. Biotechnol. 2018, 102, 9433–9448. [Google Scholar] [CrossRef] [PubMed]
- Pantou, M.P.; Kouvelis, V.N.; Typas, M.A. The complete mitochondrial genome of Fusarium oxysporum: Insights into fungal mitochondrial evolution. Gene 2008, 419, 7–15. [Google Scholar] [CrossRef]
- Torriani, S.F.; Penselin, D.; Knogge, W.; Felder, M.; Taudien, S.; Platzer, M.; McDonald, B.A.; Brunner, P.C. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet. Biol. 2014, 62, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Yang, L.; Xiang, D.; Wan, Y.; Wu, Q.; Huang, W.; Zhao, G. The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): Features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 2019, 15, 974–984. [Google Scholar] [CrossRef]
- Zaccaron, A.Z.; Bluhm, B.H. The genome sequence of Bipolaris cookei reveals mechanisms of pathogenesis underlying target leaf spot of sorghum. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Aylward, J.; Steenkamp, E.T.; Dreyer, L.L.; Roets, F.; Wingfield, B.D.; Wingfield, M.J. A plant pathology perspective of fungal genome sequencing. IMA Fungus 2017, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Franco, M.E.E.; López, S.M.Y.; Medina, R.; Lucentini, C.G.; Troncozo, M.I.; Pastorino, G.N.; Saparrat, M.C.N.; Balatti, P.A. The mitochondrial genome of the plant-pathogenic fungus Stemphylium lycopersici uncovers a dynamic structure due to repetitive and mobile elements. PLoS ONE 2017, 12, e0185545. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Li, X.; Sha, Z.; Yan, B.; Xu, Q. Complete mitochondrial genome of the Japanese snapping shrimp Alpheus japonicus (Crustacea: Decapoda: Caridea): Gene rearrangement and phylogeny within Caridea. Sci. China Life Sci. 2012, 55, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Song, S.N.; Tang, P.; Wei, S.J.; Chen, X.X. Comparative and phylogenetic analysis of the mitochondrial genomes in basal hymenopterans. Sci. Rep. 2016, 16, 20972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arfi, Y.; Buée, M.; Marchand, C.; Levasseur, A.; Record, E. Multiple markers pyrosequencing reveals highly diverse and host-specific fungal communities on the mangrove trees Avicennia marina and Rhizophora stylosa. FEMS Microbiol. Ecol. 2012, 9, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.L.; Yuan, X.L.; Du, Y.M.; Zhang, Z.F.; Zhang, P. Benzophenone Derivatives from an Algal-Endophytic Isolate of Penicillium chrysogenum and Their Cytotoxicity. Molecules 2018, 23, 3378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads—A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, 435–439. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, 54–57. [Google Scholar] [CrossRef]
- Peden, J. CodonW Version 1.4.2.; University of Nottingham: Nottingham, UK, 2005. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L. chartarum | C. trifolii | |||
|---|---|---|---|---|
| Sample ID | Contig | Scaffold | Contig | Scaffold |
| Total(bp) | 37,945,625 | 37,947,115 | 41,681,125 | 41,682,726 |
| Max(bp) | 1,026,975 | 1,409,484 | 1,476,666 | 1,819,210 |
| N50 | 234,835 | 284,119 | 400,595 | 638,944 |
| N90 | 76,366 | 94,298 | 49,549 | 65,120 |
| GC content | 50.65 % | 50.64% | 49.75 % | 49.74 % |
| Ns | 0.00% | 0.01% | 0.00% | 0.01% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.-L.; Cao, M.; Shen, G.-M.; Zhang, H.-B.; Du, Y.-M.; Zhang, Z.-F.; Li, Q.; Gao, J.-M.; Xue, L.; Wang, Z.-P.; et al. Characterization of Nuclear and Mitochondrial Genomes of Two Tobacco Endophytic Fungi Leptosphaerulina chartarum and Curvularia trifolii and Their Contributions to Phylogenetic Implications in the Pleosporales. Int. J. Mol. Sci. 2020, 21, 2461. https://doi.org/10.3390/ijms21072461
Yuan X-L, Cao M, Shen G-M, Zhang H-B, Du Y-M, Zhang Z-F, Li Q, Gao J-M, Xue L, Wang Z-P, et al. Characterization of Nuclear and Mitochondrial Genomes of Two Tobacco Endophytic Fungi Leptosphaerulina chartarum and Curvularia trifolii and Their Contributions to Phylogenetic Implications in the Pleosporales. International Journal of Molecular Sciences. 2020; 21(7):2461. https://doi.org/10.3390/ijms21072461
Chicago/Turabian StyleYuan, Xiao-Long, Min Cao, Guo-Ming Shen, Huai-Bao Zhang, Yong-Mei Du, Zhong-Feng Zhang, Qian Li, Jia-Ming Gao, Lin Xue, Zhi-Peng Wang, and et al. 2020. "Characterization of Nuclear and Mitochondrial Genomes of Two Tobacco Endophytic Fungi Leptosphaerulina chartarum and Curvularia trifolii and Their Contributions to Phylogenetic Implications in the Pleosporales" International Journal of Molecular Sciences 21, no. 7: 2461. https://doi.org/10.3390/ijms21072461