
Loss of SELENOF Induces the Transformed Phenotype in Human Immortalized Prostate Epithelial Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
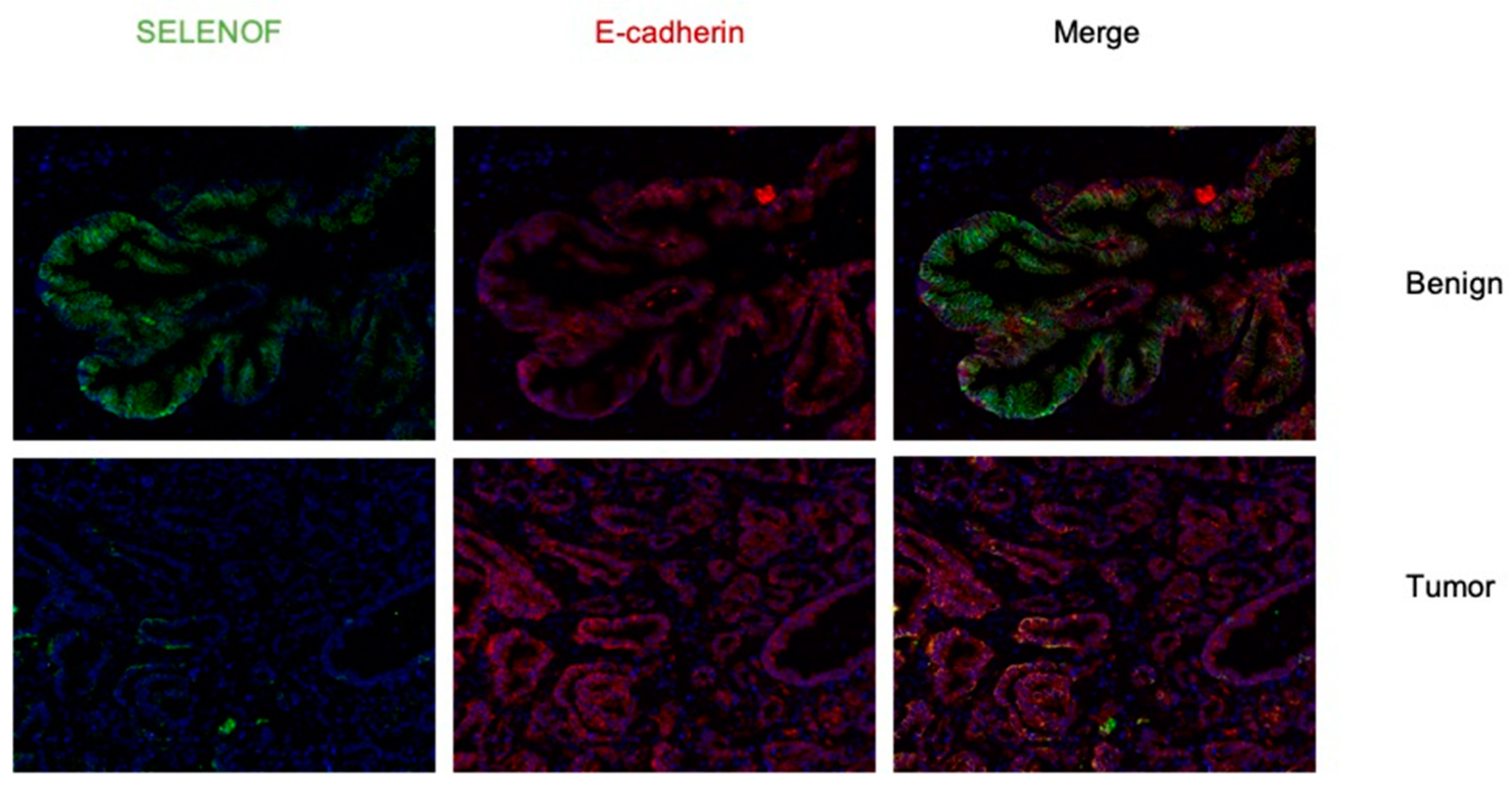
2.1. Lower Levels of SELENOF in Human Prostate Tissues Obtained from African American and Caucasian Men
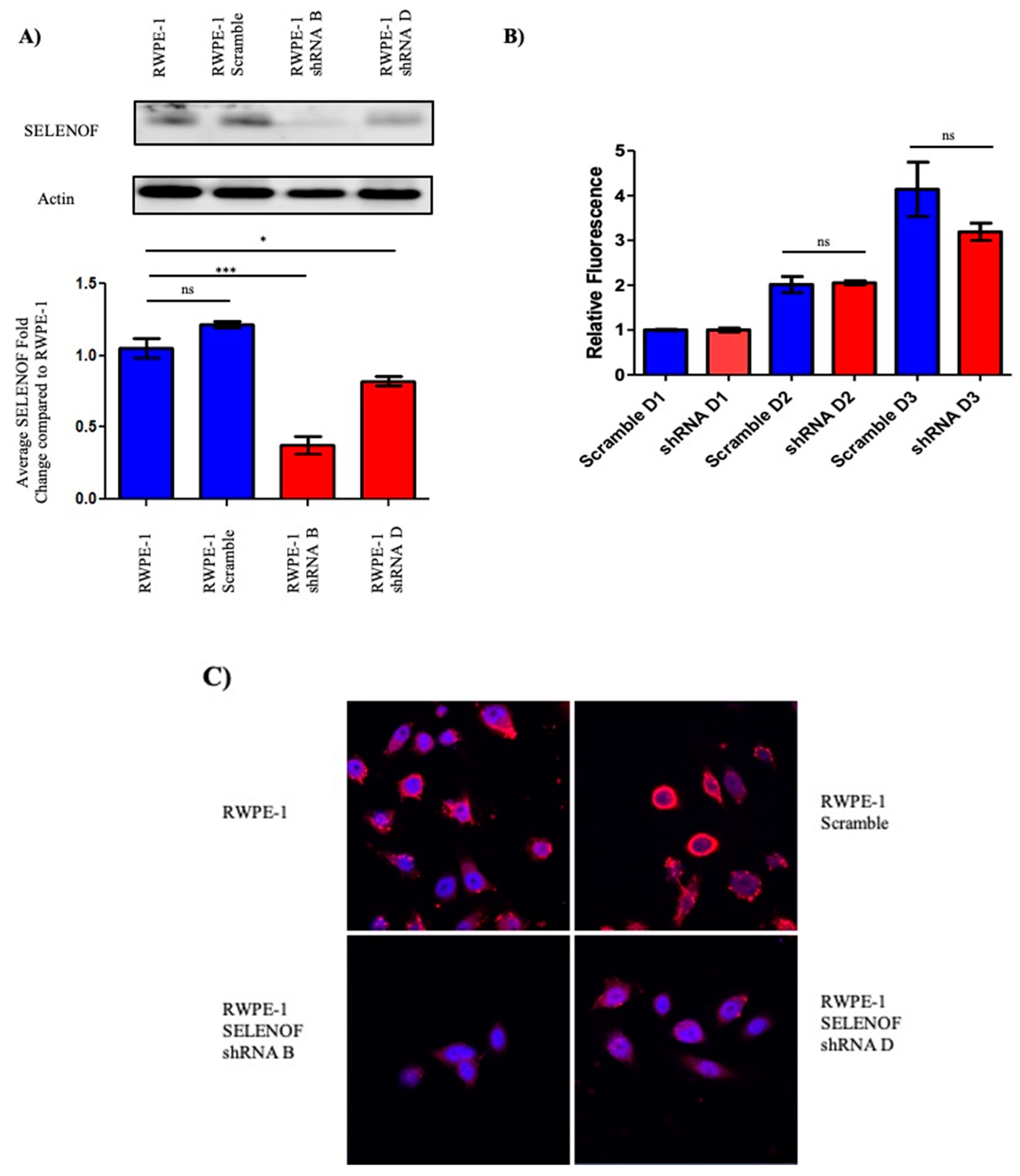
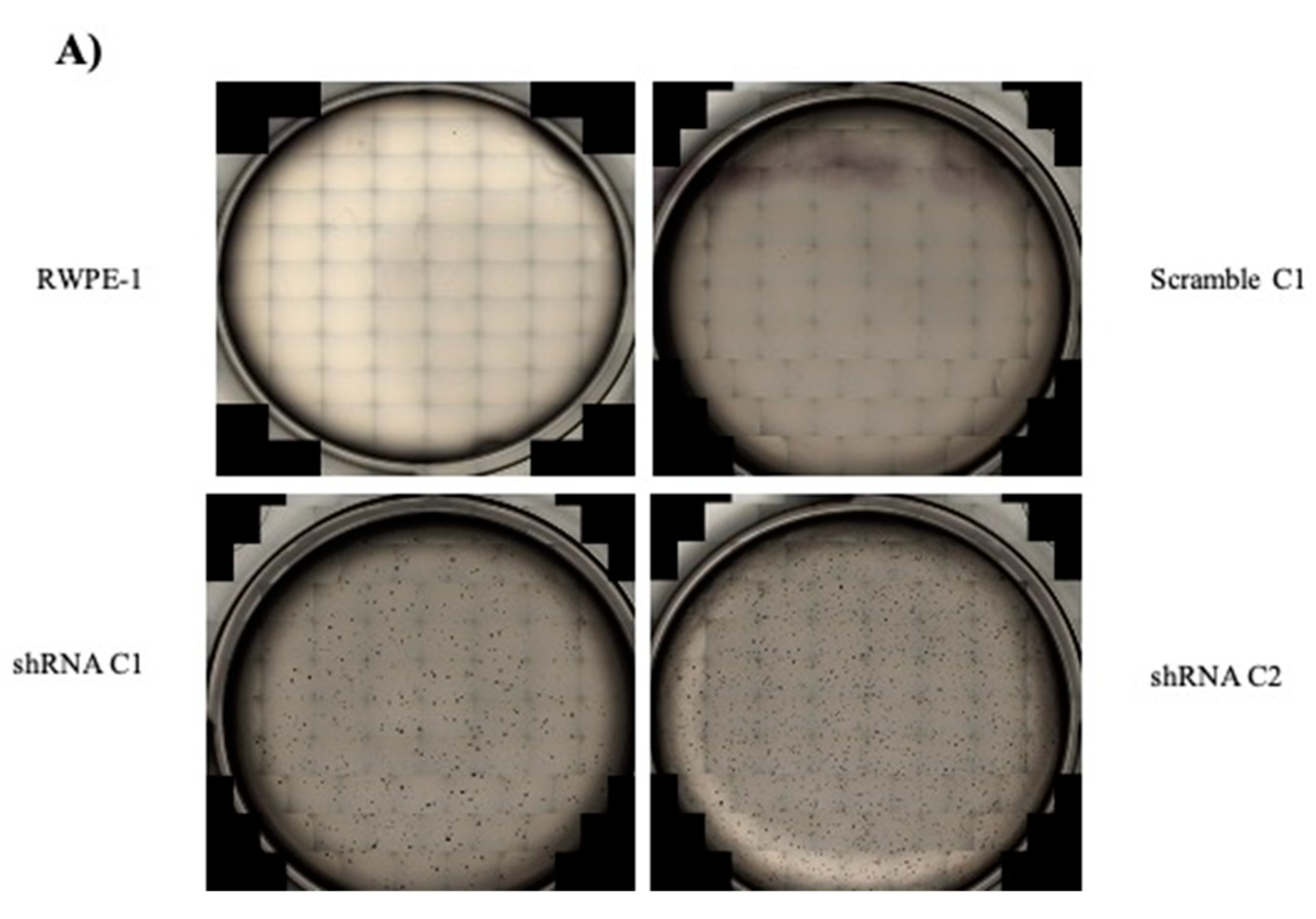
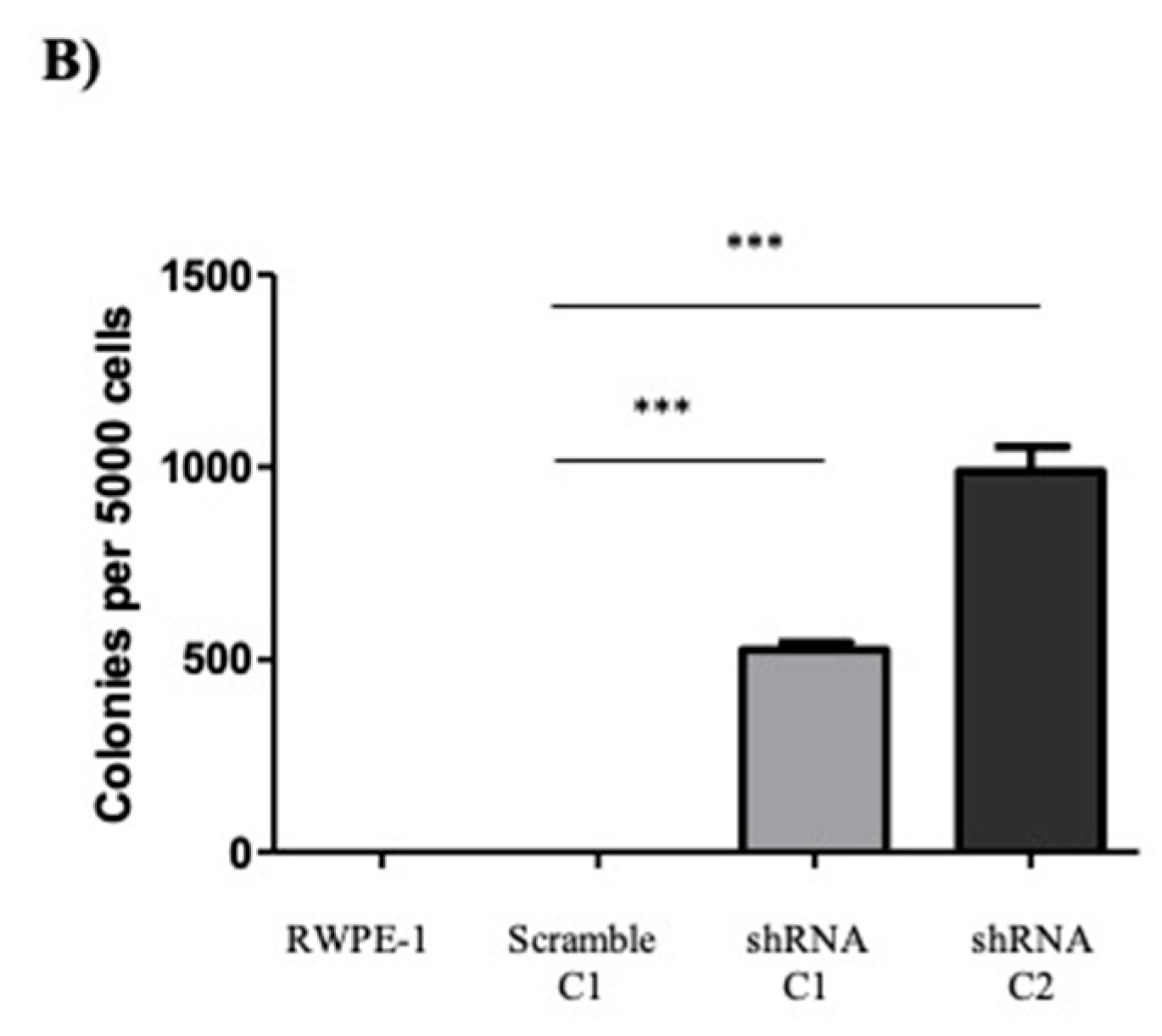
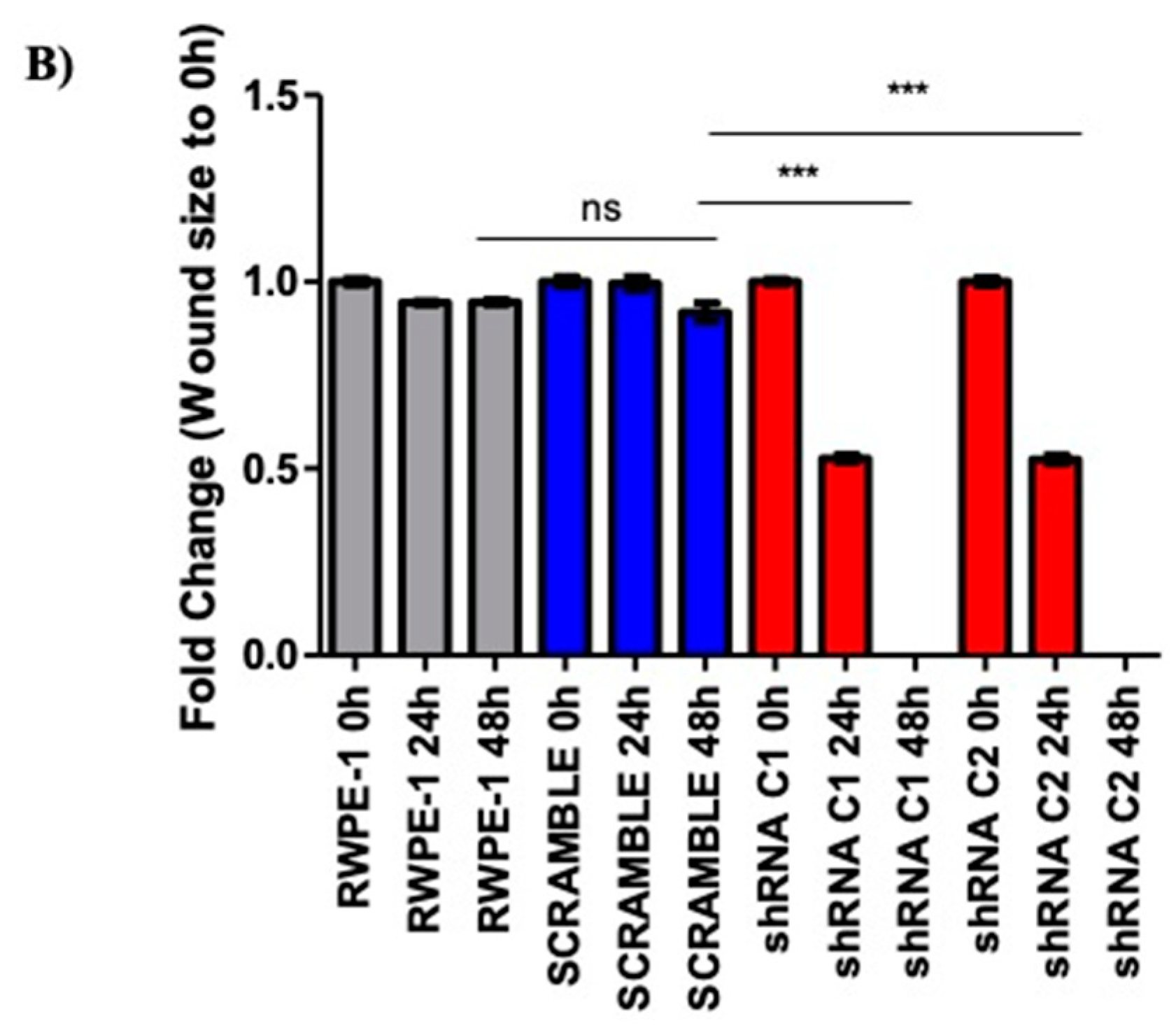
2.2. Reducing SELENOF Levels Alters Phenotypes Associated with Transformation
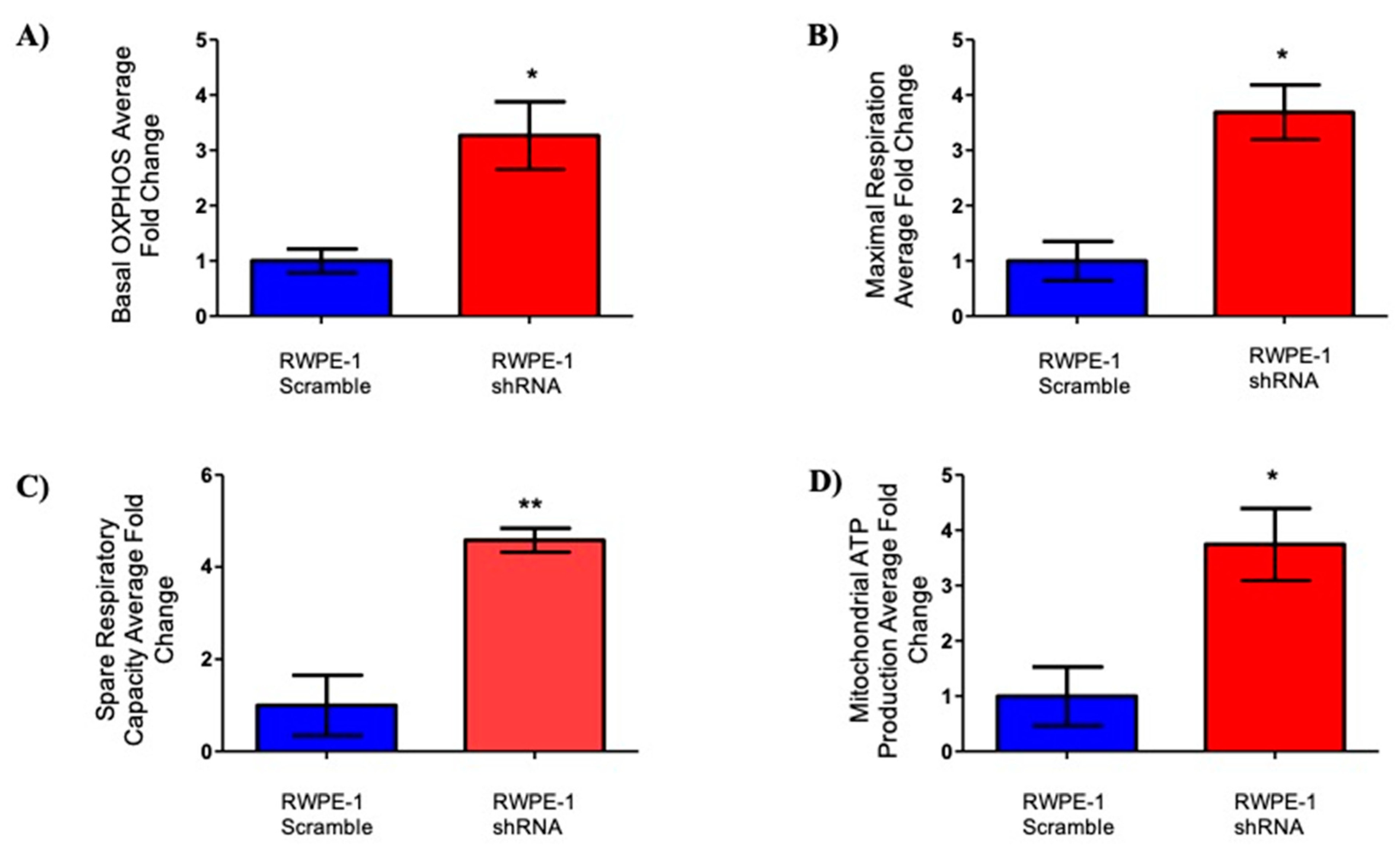
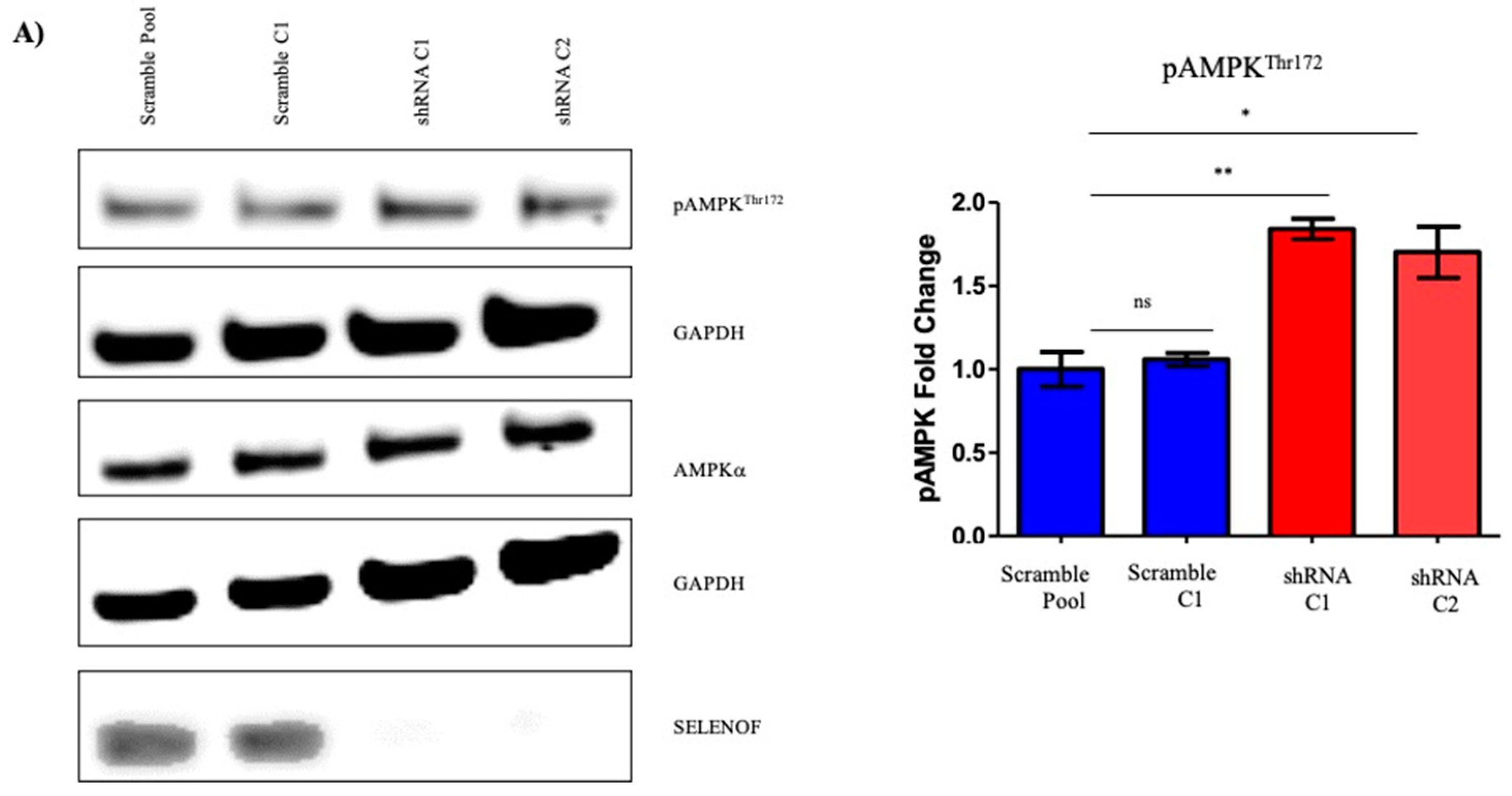
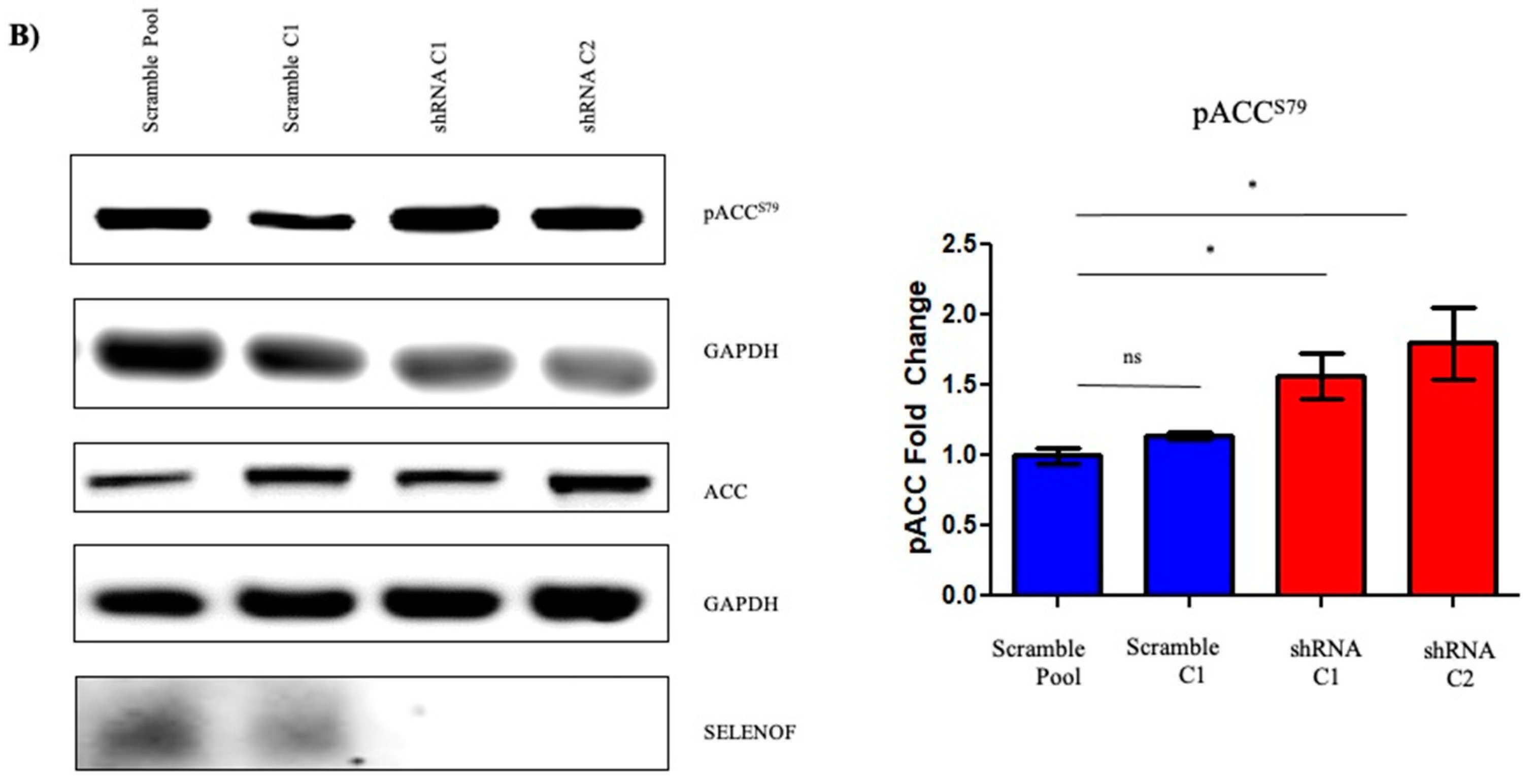
2.3. Effects of SELENOF on Metabolism
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Zeegers, M.P.; Jellema, A.; Ostrer, H. Empiric risk of prostate carcinoma for relatives of patients with prostate carcinoma: A meta-analysis. Cancer 2003, 97, 1894–1903. [Google Scholar] [CrossRef]
- Vinceti, M.; Filippini, T.; Giovane, D.C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, P.M.; Horneber, M.; D’Amico, R.; Crespi, M.C. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 1, CD005195. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Gladyshev, V.N.; Arnér, E.S.; Berry, M.J.; Brigelius-Flohé, R.; Bruford, E.A.; Burk, R.F.; Carlson, B.A.; Castellano, S.; Chavatte, L.; Conrad, M.; et al. Selenoprotein Gene Nomenclature. J. Biol. Chem. 2016, 291, 24036–24040. [Google Scholar] [CrossRef] [Green Version]
- Gladyshev, V.N.; Jeang, K.-T.; Wootton, J.C.; Hatfield, D.L. A new human selenium-containing protein: Purification, characterization and cDNA sequence. J. Biol. Chem. 1998, 273, 8910–8915. [Google Scholar] [CrossRef] [Green Version]
- Kumaraswamy, E.; Malykh, A.; Korotkov, K.V.; Hatfield, D.L.; Diamond, A.M.; Gladyshev, V.N. Structure-expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J. Biol. Chem. 2000, 275, 35540–35547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaraswamy, E.; Korotkov, K.V.; Diamond, A.M.; Gladyshev, V.N.; Hatfield, D.L. Genetic and functional analysis of mammalian Sep15 selenoprotein. Methods Enzym. 2002, 347, 187–197. [Google Scholar]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. The Sep15 protein family: Roles in disulfide bond formation and quality control in the endoplasmic reticulum. IUBMB Life 2007, 59, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Ekoue, D.N.; Ansong, E.; Liu, L.; Macias, V.; Deaton, R.; Lacher, C.; Picklo, M.; Nonn, L.; Gann, P.H.; Kajdacsy-Balla, A.; et al. Correlations of SELENOF and SELENOP genotypes with serum selenium levels and prostate cancer. Prostate 2018, 78, 279–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.J.; Korotkov, K.V.; Mehta, R.; Hatfield, D.L.; Rotimi, C.N.; Luke, A.; Prewitt, T.E.; Cooper, R.S.; Stock, W.; Vokes, E.E.; et al. Distribution and functional consequences of nucleotide polymorphisms in the 3’-untranslated region of the human Sep15 gene. Cancer Res. 2001, 61, 2307–2310. [Google Scholar] [PubMed]
- Nasr, M.; Hu, Y.J.; Diamond, A.M. Allelic loss of Sep15 in breast cancer. Cancer Ther. 2003, 1, 307–312. [Google Scholar]
- Meplan, C.; Hughes, D.J.; Pardini, B.; Naccarati, A.; Soucek, P.; Vodickova, L.; Hlavatá, I.; Vrána, D.; Vodicka, P.; Hesketh, J.E. Genetic variants in selenoprotein genes increase risk of colorectal cancer. Carcin 2010, 31, 1074–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, A.; Kim, D.H.; Relton, C.; Ahn, Y.O.; Hesketh, J. Polymorphisms in the selenoprotein S and 15-kDa selenoprotein genes are associated with altered susceptibility to colorectal cancer. Genes Nutr. 2010, 5, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Penney, K.L.; Schumacher, F.R.; Li, H.; Kraft, P.; Morris, J.S.; Kurth, T.; Mucci, L.A.; Hunter, D.J.; Kantoff, P.W.; Meir, J.; et al. A large prospective study of SEP15 genetic variation, interaction with plasma selenium levels, and prostate cancer risk and survival. Cancer Prev. Res. 2010, 3, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunasinghe, N.; Han, D.Y.; Zhu, S.; Yu, J.; Lange, K.; Duan, H.; Medhora, R.; Singh, N.; Kan, J.; Alzaher, W.; et al. Serum selenium and single-nucleotide polymorphisms in genes for selenoproteins: Relationship to markers of oxidative stress in men from Auckland, New Zealand. Genes Nutr. 2012, 7, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbrecher, A.; Méplan, C.; Hesketh, J.; Schomburg, L.; Endermann, T.; Jansen, E.; Åkesson, B.; Rohrmann, S.; Linseisen, J. Effects of selenium status and polymorphisms in selenoprotein genes on prostate cancer risk in a prospective study of European men. Cancer Epidem. Bio. Prev. 2010, 19, 2958–2968. [Google Scholar] [CrossRef] [Green Version]
- Gleason, D.F.; Mellinger, G.T. Prediction of prognosis for prostatic adenocarcinoma by combined histological grading and clinical staging. J. Urol. 1974, 111, 58–64. [Google Scholar] [CrossRef]
- Epstein, J.I.; Allsbrook, W.C., Jr.; Amin, M.B.; Egevad, L.L.; Committee, I.G. The 2005 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Am. J. Surg. Pathol. 2005, 29, 1228–1242. [Google Scholar] [CrossRef] [Green Version]
- Borowicz, S.; Scoyk, M.V.; Avasarala, S.; Rathinam, M.K.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef] [Green Version]
- Puck, T.T.; Marcus, P.I.; Cieciura, S.J. Clonal growth of mammalian cells in vitro; growth characteristics of colonies from single HeLa cells with and without a feeder layer. J. Exp. Med. 1956, 103, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Todaro, G.J.; Lazar, G.K.; Green, H. The initiation of cell division in a contact-inhibited mammalian cell line. J. Cell. Comp. Physiol. 1965, 66, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Cory, G. Scratch-wound Assay. Meth. Mol. Biol. 2011, 769, 25–30. [Google Scholar]
- Costello, L.C.; Franklin, R.B.; Feng, P.; Tan, M.; Bagasra, O. Zinc and prostate cancer: A critical scientific, medical, and public interest issue (United States). Cancer Causes Control. 2005, 16, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Cutruzzola, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose Metabolism in the Progression of Prostate Cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, L.C.; Franklin, R.B. The intermediary metabolism of the prostate: A key to understanding the pathogenesis and progression of prostate malignancy. Oncology 2000, 59, 269–282. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Ren, B.; Wang, H.; Huang, R.; Zhou, J.; Liu, H.; Tian, J.; Huang, K. Hepatic proteomic analysis of selenoprotein F knockout mice by iTRAQ: An implication for the roles of selenoprotein F in metabolism and diseases. J. Proteom. 2020, 215, 103653. [Google Scholar] [CrossRef]
- Zheng, X.; Ren, B.; Wang, H.; Huang, R.; Zhou, J.; Liu, H.; Tian, J.; Huang, K. Selenoprotein F knockout leads to glucose and lipid metabolism disorders in mice. J. Biol. Inorg. Chem. 2020, 25, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, J.; Zhang, N.; Yang, Q.; Jin, Y.; Wang, Y. The acetyl-CoA carboxylase enzyme: A target for cancer therapy? Expert Rev. Anticancer Ther. 2015, 15, 667–676. [Google Scholar] [CrossRef]
- Yim, S.H.; Everley, R.A.; Schildberg, F.A.; Lee, S.-G.; Orsi, A.; Barbati, Z.R.; Fomenko, D.E.; Tsuji, P.A.; Luo, H.R.; Gygi, S.P.; et al. Role of Selenof as a Gatekeeper of Secreted Disulfide-Rich Glycoproteins. Cell Rep. 2018, 23, 1387–1398. [Google Scholar] [CrossRef] [Green Version]
- Xun, P.; Bujnowski, D.; Liu, K.; Morris, J.S.; Guo, Z.; He, K. Distribution of toenail selenium levels in young adult Caucasians and African Americans in the United States: The CARDIA Trace Element Study. Environ. Res. 2011, 111, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield-Lillico, A.J.; Slate, E.H.; Reid, M.E.; Turnbull, B.W.; Wilkins, P.A.; Combs, G.F., Jr.; Park, H.K.; Gross, E.G.; Graham, G.F.; Stratton, M.S.; et al. Selenium supplementation and secondary prevention of nonmelanoma skin cancer in a randomized trial. J. Natl. Cancer Inst. 2003, 95, 1477–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenfield, S.A.; Blarigan, E.L.V.; DuPre, N.; Stampfer, M.J.; Giovannucci, E.L.; Chan, J.M. Selenium supplementation and prostate cancer risk. J. Natl. Cancer Inst. 2015, 107, 360–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinceti, M.; Filipini, T.; Rothman, K.J. Selenium exposure and the risk of type 2 diabetes: A systematic review and meta-analysis. Eur. J. Epidemiol. 2018, 33, 789–810. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, L.K.; Kadkol, S.; Sverdlov, M.; Kastrati, I.; Elhodaky, M.; Deaton, R.; Sfanos, K.S.; Wang, H.; Liu, L.; Diamond, A.M. Loss of SELENOF Induces the Transformed Phenotype in Human Immortalized Prostate Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 12040. https://doi.org/10.3390/ijms222112040
Hong LK, Kadkol S, Sverdlov M, Kastrati I, Elhodaky M, Deaton R, Sfanos KS, Wang H, Liu L, Diamond AM. Loss of SELENOF Induces the Transformed Phenotype in Human Immortalized Prostate Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(21):12040. https://doi.org/10.3390/ijms222112040
Chicago/Turabian StyleHong, Lenny K., Shrinidhi Kadkol, Maria Sverdlov, Irida Kastrati, Mostafa Elhodaky, Ryan Deaton, Karen S. Sfanos, Heidi Wang, Li Liu, and Alan M. Diamond. 2021. "Loss of SELENOF Induces the Transformed Phenotype in Human Immortalized Prostate Epithelial Cells" International Journal of Molecular Sciences 22, no. 21: 12040. https://doi.org/10.3390/ijms222112040