
Characteristics of the Complete Plastid Genome Sequences of the Monotypic Genus Dodecadenia (Family: Lauraceae) and Its Phylogenomic Implications

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Plastome Sequencing
2.2. Plastome Assembly and Annotation
2.3. Sequence Analysis
2.4. Phylogenetic Analysis
2.5. Sequence Divergence and Comparative Genome Analysis
3. Results
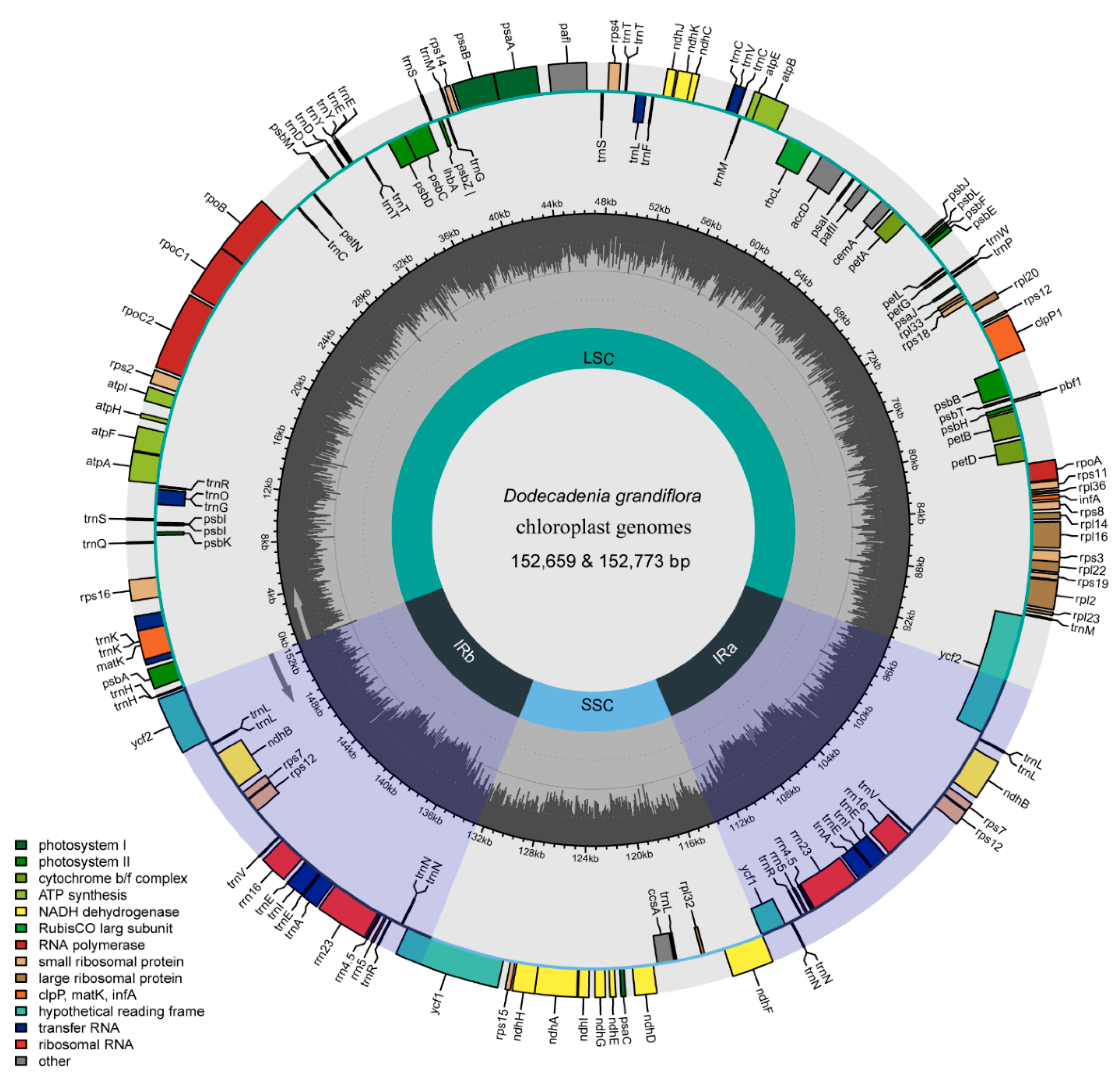
3.1. General Feature of the Plastome
3.2. Amino Acid Frequency and Codon Usage Analysis
3.3. RNA Editing Sites Analysis
3.4. Repeats Analysis
3.5. Phylogenetic Analyses
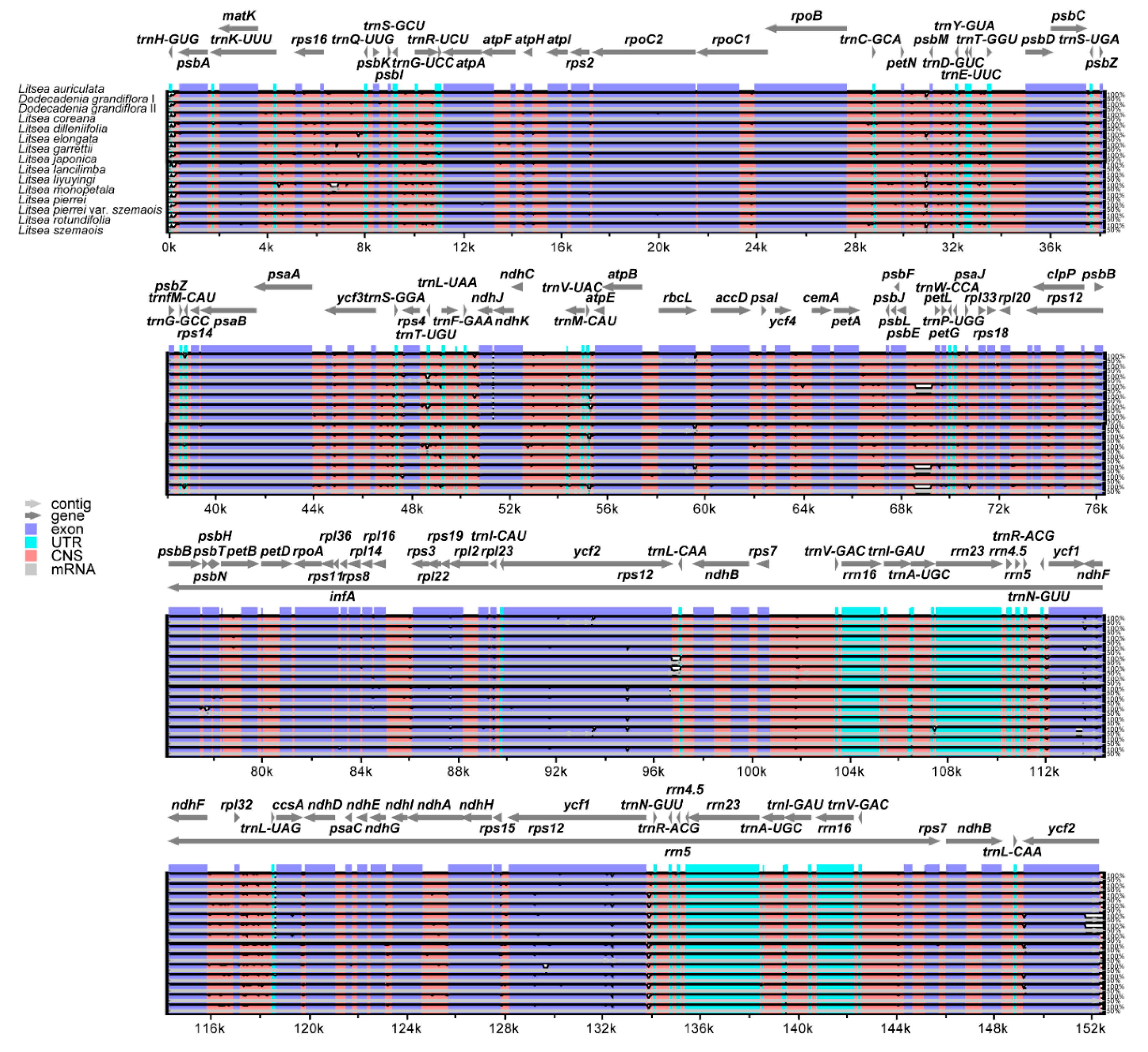
3.6. Comparative Plastome Sequence Divergence and Hotspots Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rohwer, J.G. Lauraceae. In Flowering Plants Dicotyledons; Springer: Berlin/Heidelberg, Germany, 1993; pp. 366–391. [Google Scholar]
- Li, J.; Christophel, D.C. Systematic relationships within the Litsea complex (Lauraceae): A cladistic analysis on the basis of morphological and leaf cuticle data. Aust. Syst. Bot. 2000, 13, 1–13. [Google Scholar] [CrossRef]
- Chanderbali, A.S.; Van Der Werff, H.; Renner, S.S. Phylogeny and historical biogeography of Lauraceae: Evidence from the chloroplast and nuclear genomes. Ann. Mo. Bot. Gard. 2001, 88, 104–134. [Google Scholar] [CrossRef] [Green Version]
- Fijridiyanto, I.A.; Murakami, N. Molecular systematics of Malesian Litsea Lam. and putative related genera (Lauraceae). Acta Phytotax. Geobot. 2009, 60, 1–18. [Google Scholar] [CrossRef]
- Jo, S.; Kim, Y.K.; Cheon, S.H.; Fan, Q.; Kim, K.J. Characterization of 20 complete plastomes from the tribe Laureae (Lauraceae) and distribution of small inversions. PLoS ONE 2019, 14, e0224622. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yu, W.B.; Tan, Y.H.; Jin, J.J.; Wang, B.; Yang, J.B.; Liu, B.; Corlett, R.T. Plastid phylogenomics improve phylogenetic resolution in the Lauraceae. J. Syst. Evol. 2020, 58, 423–439. [Google Scholar] [CrossRef]
- Xiao, T.W.; Xu, Y.; Jin, L.; Liu, T.J.; Yan, H.F.; Ge, X.J. Conflicting phylogenetic signals in plastomes of the tribe Laureae (Lauraceae). PeerJ 2020, 8, e10155. [Google Scholar] [CrossRef] [PubMed]
- Van Der Werff, H.; Richter, V.D.H. Toward an improved classification of Lauraceae. Ann. Mo. Bot. Gard. 1996, 83, 409–418. [Google Scholar] [CrossRef]
- Kostermans, A.J.G.H. Lauraceae. Reinwardtia 1957, 4, 193–256. [Google Scholar]
- Tian, X.; Ye, J.; Song, Y. Plastome sequences help to improve the systematic position of trinerved Lindera species in the family Lauraceae. PeerJ 2019, 7, e7662. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Christophel, D.C.; Conran, J.G.; Li, H.W. Phylogenetic relationships within the ‘core’ Laureae (Litsea complex, Lauraceae) inferred from sequences of the chloroplast gene matK and nuclear ribosomal DNA ITS regions. Plant Syst. Evol. 2004, 246, 19–34. [Google Scholar] [CrossRef]
- Li, J.; Conran, J.G.; Christophel, D.C.; Li, Z.M.; Li, L.; Li, H.W. Phylogenetic relationships of the Litsea complex and core Laureae (Lauraceae) using ITS and ETS sequences and morphology. Ann. Mo. Bot. Gard. 2008, 95, 580–599. [Google Scholar] [CrossRef]
- Liu, Z.F.; Ma, H.; Ci, X.Q.; Li, L.; Song, Y.; Liu, B.; Li, H.W.; Wang, S.L.; Qu, X.J.; Hu, J.L. Can plastid genome sequencing be used for species identification in Lauraceae? Bot. J. Linn. Soc. 2021, 197, 1–14. [Google Scholar] [CrossRef]
- Han, C.; Ding, R.; Zong, X.; Zhang, L.; Chen, X.; Qu, B. Structural characterization of Platanthera ussuriensis chloroplast genome and comparative analyses with other species of Orchidaceae. BMC Genom. 2022, 23, 1–13. [Google Scholar] [CrossRef]
- Xiao, T.W.; Yan, H.F.; Ge, X.J. Plastid phylogenomics of tribe Perseeae (Lauraceae) yields insights into the evolution of East Asian subtropical evergreen broad-leaved forests. BMC Plant Biol. 2022, 22, 1–15. [Google Scholar] [CrossRef]
- Zong, D.; Gan, P.; Zhou, A.; Zhang, Y.; Zou, X.; Duan, A.; Song, Y.; He, C. Plastome sequences help to resolve deep-level relationships of Populus in the family Salicaceae. Front. Plant Sci. 2019, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.S.; Wang, Y.L.; Xia, N.H.; Liu, Y.; Liu, M.; Lian, L.; Li, N.; Li, L.F.; Lang, X.A.; Gong, Y.Q.; et al. Plastid and nuclear phylogenomic incongruences and biogeographic implications of Magnolia s.l. (Magnoliaceae). J. Syst. Evol. 2022, 161, 107171. [Google Scholar] [CrossRef]
- Yu, J.; Fu, J.; Fang, Y.; Xiang, J.; Dong, H. Complete chloroplast genomes of Rubus species (Rosaceae) and comparative analysis within the genus. BMC Genom. 2022, 23, 1–14. [Google Scholar] [CrossRef]
- Liu, C.; Chen, H.; Tang, L.; Khine, P.K.; Han, L.; Song, Y.; Tan, Y. Plastid genome evolution of a monophyletic group in the subtribe Lauriineae (Laureae, Lauraceae). Plant Divers. 2022, 44, 377–388. [Google Scholar] [CrossRef]
- Trofimov, D.; Cadar, D.; Schmidt-Chanasit, J.; Rodrigues De Moraes, P.L.; Rohwer, J.G. A comparative analysis of complete chloroplast genomes of seven Ocotea species (Lauraceae) confirms low sequence divergence within the Ocotea complex. Sci. Rep. 2022, 12, 1120. [Google Scholar] [CrossRef]
- Kumar, M.; Rawat, P.; Rahuja, N.; Srivastava, A.K.; Maurya, R. Antihyperglycemic activity of phenylpropanoyl esters of catechol glycoside and its dimers from Dodecadenia grandiflora. Phytochemistry 2009, 70, 1448–1455. [Google Scholar] [CrossRef]
- Doyle, J.J.; Dickson, E.E. Preservation of plant samples for DNA restriction endonuclease analysis. Taxon 1987, 36, 715–722. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; Depamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Zheng, S.; Poczai, P.; Hyvönen, J.; Tang, J.; Amiryousefi, A. Chloroplot: An online program for the versatile plotting of organelle genomes. Front. Genet. 2020, 11, 1123. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Delbarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, Y.; Liu, Y.; Yang, L.; Dong, R.; Jiang, L.; Liu, P.; Liu, G.; Wang, Z.; Luo, L. Genome-wide analysis of tandem duplicated genes and their contribution to stress resistance in pigeonpea (Cajanus cajan). Genomics 2021, 113, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, Y.; Tng, D.Y.P.; Zhou, J.; Zhang, Y.; Wang, Z.; Li, P.; Wang, Z. Comparative chloroplast genomics of Litsea Lam. (Lauraceae) and its phylogenetic implications. Forests 2021, 12, 744. [Google Scholar] [CrossRef]
- Song, Y.; Yao, X.; Tan, Y.; Gan, Y.; Yang, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two subtropical trees, Phoebe sheareri and Phoebe omeiensis (Lauraceae). Tree Genet. Genom. 2017, 13, 120. [Google Scholar] [CrossRef]
- Song, Y.; Yu, W.B.; Tan, Y.; Liu, B.; Yao, X.; Jin, J.; Padmanaba, M.; Yang, J.B.; Corlett, R.T. Evolutionary comparisons of the chloroplast genome in Lauraceae and insights into loss events in the Magnoliids. Genome Biol. Evol. 2017, 9, 2354–2364. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Yao, X.; Liu, B.; Tan, Y.; Corlett, R.T. Complete plastid genome sequences of three tropical Alseodaphne trees in the family Lauraceae. Holzforschung 2018, 72, 337–345. [Google Scholar] [CrossRef]
- Zhao, M.L.; Song, Y.; Ni, J.; Yao, X.; Tan, Y.H.; Xu, Z.F. Comparative chloroplast genomics and phylogenetics of nine Lindera species (Lauraceae). Sci. Rep. 2018, 8, 8844. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Chen, H.; Cai, J.; Han, L. The complete plastid genome of a medicinal tree Lindera chienii Cheng 1934 (Lauraceae: Laureae). Mitochondrial DNA B 2022, 7, 1252–1254. [Google Scholar] [CrossRef]
- Liu, C.; Chen, H.; Han, L.; Tang, L. The complete plastid genome of an evergreen tree Litsea elongata (Lauraceae: Laureae). Mitochondrial DNA B 2020, 5, 2483–2484. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, B.; Davis, C.C.; Yang, Y. Plastome phylogenomics, systematics, and divergence time estimation of the Beilschmiedia group (Lauraceae). Mol. Phylogenet. Evol. 2020, 151, 106901. [Google Scholar] [CrossRef]
- Zhu, B.; Qian, F.; Hou, Y.; Yang, W.; Cai, M.; Wu, X. Complete chloroplast genome features and phylogenetic analysis of Eruca sativa (Brassicaceae). PLoS ONE 2021, 16, e0248556. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, Y.; Yang, G.; Peng, J.; Chen, S.; Xu, Z. Determination of the evolutionary pressure on Camellia oleifera on Hainan Island using the complete chloroplast genome sequence. PeerJ 2019, 7, e7210. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.L.; Wang, R.N.; Zhang, N.Y.; Fan, W.B.; Fang, M.F.; Li, Z.H. Molecular evolution of chloroplast genomes of orchid species: Insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 716. [Google Scholar] [CrossRef] [Green Version]
- Mehmetoglu, E.; Kaymaz, Y.; Ates, D.; Kahraman, A.; Tanyolac, M.B. The complete chloroplast genome sequence of Cicer echinospermum, genome organization and comparison with related species. Sci. Hortic. 2022, 296, 110912. [Google Scholar] [CrossRef]
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Chen, H.; Cai, J.; Tian, X.; Han, L.; Song, Y. Characteristics of the Complete Plastid Genome Sequences of the Monotypic Genus Dodecadenia (Family: Lauraceae) and Its Phylogenomic Implications. Forests 2022, 13, 1240. https://doi.org/10.3390/f13081240
Liu C, Chen H, Cai J, Tian X, Han L, Song Y. Characteristics of the Complete Plastid Genome Sequences of the Monotypic Genus Dodecadenia (Family: Lauraceae) and Its Phylogenomic Implications. Forests. 2022; 13(8):1240. https://doi.org/10.3390/f13081240
Chicago/Turabian StyleLiu, Chao, Huanhuan Chen, Jian Cai, Xiangyu Tian, Lihong Han, and Yu Song. 2022. "Characteristics of the Complete Plastid Genome Sequences of the Monotypic Genus Dodecadenia (Family: Lauraceae) and Its Phylogenomic Implications" Forests 13, no. 8: 1240. https://doi.org/10.3390/f13081240