
Chromatin Regulator SPEN/SHARP in X Inactivation and Disease
by
 ,
,
Benedetto Daniele Giaimo
1,*,
Teresa Robert-Finestra
2,
Franz Oswald
3,
Joost Gribnau
2 and
Tilman Borggrefe
1,* 1
Institute of Biochemistry, University of Giessen, Friedrichstrasse 24, 35392 Giessen, Germany
2
Department of Developmental Biology, Erasmus MC, Oncode Institute, Wytemaweg 80, 3015 CN Rotterdam, The Netherlands
3
Center for Internal Medicine, Department of Internal Medicine I, University Medical Center Ulm, Albert-Einstein-Allee 23, 89081 Ulm, Germany
*
Authors to whom correspondence should be addressed.
Cancers 2021, 13(7), 1665; https://doi.org/10.3390/cancers13071665
Submission received: 23 February 2021
/
Revised: 26 March 2021
/
Accepted: 26 March 2021
/
Published: 1 April 2021
(This article belongs to the Section Molecular Cancer Biology)
Abstract
:Simple Summary
Carcinogenesis is a multistep process involving not only the activation of oncogenes and disabling tumor suppressor genes, but also epigenetic modulation of gene expression. X chromosome inactivation (XCI) is a paradigm to study heterochromatin formation and maintenance. The double dosage of X chromosomal genes in female mammals is incompatible with early development. XCI is an excellent model system for understanding the establishment of facultative heterochromatin initiated by the expression of a 17,000 nt long non-coding RNA, known as X inactive specific transcript (Xist), on the X chromosome. This review focuses on the molecular mechanisms of how epigenetic modulators act in a step-wise manner to establish facultative heterochromatin, and we put these in the context of cancer biology and disease. An in depth understanding of XCI will allow a better characterization of particular types of cancer and hopefully facilitate the development of novel epigenetic therapies.
Abstract
Enzymes, such as histone methyltransferases and demethylases, histone acetyltransferases and deacetylases, and DNA methyltransferases are known as epigenetic modifiers that are often implicated in tumorigenesis and disease. One of the best-studied chromatin-based mechanism is X chromosome inactivation (XCI), a process that establishes facultative heterochromatin on only one X chromosome in females and establishes the right dosage of gene expression. The specificity factor for this process is the long non-coding RNA X inactive specific transcript (Xist), which is upregulated from one X chromosome in female cells. Subsequently, Xist is bound by the corepressor SHARP/SPEN, recruiting and/or activating histone deacetylases (HDACs), leading to the loss of active chromatin marks such as H3K27ac. In addition, polycomb complexes PRC1 and PRC2 establish wide-spread accumulation of H3K27me3 and H2AK119ub1 chromatin marks. The lack of active marks and establishment of repressive marks set the stage for DNA methyltransferases (DNMTs) to stably silence the X chromosome. Here, we will review the recent advances in understanding the molecular mechanisms of how heterochromatin formation is established and put this into the context of carcinogenesis and disease.
Keywords:
XCI; SHARP; Spen; NCoR; HDAC; polycomb; DNA methylation; transcription; silencing; repression1. Long Non-Coding RNAs and Cancer
Less than 2% of the genome is transcribed in protein-encoding mRNAs; however, most of it is actively transcribed, which suggests that a fraction produces non-coding RNAs (ncRNAs). ncRNAs are classified based on their size in small ncRNAs (<200 bp) and long ncRNAs (>200 bp, also referred to as lncRNAs) [1,2]. In this review, we focus on lncRNAs.
lncRNAs can be classified based on their genomic localization [3] as well as on their cellular distribution [4]. It is proposed that lncRNAs are organized in secondary and tertiary structures [5] that may offer binding surfaces for proteins containing RNA-recognition motives (RRMs). lncRNAs are capable of interacting with coactivators or corepressors of transcription, recruiting them to specific genes or genomic regions [6,7,8,9]. In addition, lncRNAs are also able to regulate alternative splicing events by interacting with splicing factors [6,10].
Several lncRNAs have been associated with variety of diseases. Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was found to be upregulated in renal cell carcinoma (RCC), gastric cancer (GC), gallbladder cancer (GBC), colorectal cancer (CRC), multiple myeloma, clear cell renal cell carcinoma (ccRCC), and glioma, as well as in osteosarcoma [11,12,13,14,15,16,17,18], and it has been proposed as a molecular marker therein [14,15,16,19]. The lncRNA imprinted H19 gene is maternally expressed and strongly downregulated directly after birth [20,21,22]. It was shown that H19 is strongly upregulated in gastric cancer [23,24,25], similarly to several other lncRNAs, such as PVT1 oncogene (PVT1), gastric carcinoma high expressed transcript 1 (GHET1), antisense ncRNA in the INK4 locus (ANRIL), SPRY4 intronic transcript 1 (SPRY4-IT1), and the already mentioned MALAT1 [18,26,27,28,29,30]. H19 is also upregulated in other cancer types, such as esophageal cancer, CRC and lung cancer [25]. Another example is represented by homeobox (HOX) transcript antisense RNA (HOTAIR), which is upregulated in hepatocellular carcinoma [31], in colorectal cancer [32], in gastric cancer [33], and pancreatic cancer [34].
In this review, we will focus our attention on X inactive specific transcript (Xist; XIST in human), a lncRNA whose main function is to inactivate one X chromosome in female cells to achieve dosage compensation between males (XY) and females (XX) (see below). Recent studies highlighted its frequent deregulation in cancer. XIST is responsible for silencing several genes, and the observation that the X-linked oncogenes ARAF-1 and ETS-like 1 (ELK-1) are overexpressed in tumors with multiple active X chromosomes [35] suggests that the deregulation of XIST may be associated with cancer. Several studies observed defective X chromosome inactivation (XCI) in breast and basal-like cancer and linked the deregulation of the X chromosome to breast cancer (BC) [36,37,38,39,40,41,42], to ovarian cancer [43], as well as to cancers in patients affected by Klinefelter syndrome [44]. This deregulation is usually given by a loss of XIST as result of disappearance of the inactive X chromosome (Xi) and amplification of the active one (Xa) [37,38,40,43,44].
The gathered knowledge of these studies suggest that lncRNAs are important mediators of pathological conditions and they may, in the future, serve as potential therapeutic targets.
XCI serves as a powerful paradigm to study chromatin dynamics at a chromosomal scale. XCI co-evolved with the mammalian sex chromosomes as a mechanism to equalize the dosage of X-encoded genes between male XY and female XX cells. The central player in this process is Xist, which was discovered as the first functional lncRNA in mammals, being upregulated from the future Xi, coating the Xi in cis, thereby recruiting chromatin remodelers directly and indirectly rendering the X chromosome inactive. Xist is located on the X chromosome and it is surrounded by several other lncRNA-encoding genes, including Tsix, just proximal to Xist (Jpx), and five prime to XistT (Ftx), which, in mouse, have been shown to be involved in Xist regulation through different mechanisms, including transcriptional interference, RNA-mediated recruitment of chromatin remodelers, and through transcription co-activation [45,46,47,48]. Xist encodes a 17 kb lncRNA (19 kb in human) that contains six repeat structures that play a crucial, sometimes redundant, role in Xist-mediated silencing as well as localization [49]. So far, most of the functional studies have been performed in mouse where deletions of the most 5′ located repeat A led to a silencing phenotype despite the fact that Xist spreading was unaffected. Several studies indicated that SHARP [SMRT (silencing mediator for retinoid or thyroid hormone receptors) and HDACs (histone deacetylases)-associated repressor protein], encoded by the SPEN (split ends) gene [also called SHARP or Mint (Msx2-interacting nuclear target protein)], is a crucial factor in the X inactivation process through interacting with the A repeat sequence and recruitment of several repressor complex members, such as nuclear receptor corepressor (NCoR), SMRT, and nucleosome remodeling deacetylase (NuRD) complexes [50,51,52,53,54,55] (see Table 1).
SHARP is transiently enriched at the promoters and enhancers of genes that are subject to XCI and it recruits NCoR/SMRT complexes that contain HDACs, leading to histone deacetylation [55]. SHARP localization also shows overlap with NuRD complex members predominantly at promoters, and its action is only required during the initiation phase of XCI, as removal of SHARP after Xi is established has no effect [55,140]). As a consequence of the action of SHARP and its associated protein complexes, promoters and enhancers are deacetylated in a stepwise manner, paving the way for the action of the polycomb group (PcG) protein repressive complexes PRC1 and PRC2 that play a crucial role in the establishment and maintenance of the silent state of the Xi. PRC1 is a large multi-protein complex that is recruited to Xist through heterogeneous nuclear ribonucleoprotein K (hnRNPK) that acts as a bridge between PRC1 and Xist Repeat B and, to a lesser extent, Repeat C [65,66,77]. PRC1-directed deposition of monoubiquitination of K119 of histone H2A (H2A119ub1) is mediated by the core PRC1 complex member really interesting new gene 1 isoform A or B (RING1A/B) and, in turn, is recognized by PRC2 subunit jumanji and AT-rich interaction domain-containing 2 (JARID2) facilitating trimethylation on K27 of histone H3 (H3K27me3) by the enhancer of zeste 2/lysine (K) methyltransferase 6A (EZH2/KMT6A) [141,142,143]. Subsequently, PRC1 and PRC2 recruitment is re-enforced through the recruitment of PRC1 that recognizes the trimethylation of K27 of histone H3 (H3K27me3) through chromobox-containing protein (CBX), which further promotes H2AK119ub1 deposition, facilitating the spreading of silencing [144,145,146]. At a later stage of the XCI process, de novo DNA methyltransferases (DNMTs) are recruited to lock in the silent state through the deposition of DNA methylation at promoters and CpG islands (CGI). These studies highlight the concerted action of chromatin readers and writers directing the right order of epigenetic events required to establish the Xi that is propagated through a near infinite number of cell divisions.
2. The Inactive X Chromosome Status in Cancer
The complete loss or alteration of the Xi is frequently observed in breast and ovarian cancers, amongst other types of cancer [147,148]. Initial studies showed that Xist/XIST RNA is essential for the initiation and establishment of XCI during development, but dispensable to maintain the Xi in female somatic cells [149,150]. Even so, more recent studies making use of more sensitive techniques detect the reactivation of X-linked genes upon nearly complete or partial Xist/XIST depletion. The human X chromosome codes for more than 900 coding genes [151], including several tumor suppressor genes and oncogenes [152,153]. Thus, gene dosage changes that are caused by potential reactivation or silencing of X-linked genes could be detrimental. So far, only one well documented study in mice revealed a clear causal relationship between Xist deletion in the hematopoietic lineage and high penetrance hematopoietic cancer [154].
In human, the absence of the Xi (Barr body) in female cancer cells and presence of multiple Xa’s have been frequently associated with different forms of cancer, such as breast cancer [38,40,44]. However, these events are primarily attributed to the loss of the Xi and duplication of the Xa due to chromosome segregation errors (see Figure 1) [38,40,44].
Epigenetic alterations that are caused by epigenetic erosion of the Xi have also been described. These erosion events affect histone modification, deposition, and DNA methylation, leading to the reactivation of X-linked genes in breast cancer cell lines and primary tumors [155]. Moreover, the Xi in female cancer genomes has been shown to accumulate more mutations than the autosomes in various cancer types, including medulloblastoma, breast cancer, glioblastoma, and acute myeloid leukemia (AML) [156]. Interestingly, recent studies suggest that high XIST expression levels correlate with a poor survival in various types of cancer [157]. Some of these studies propose that XIST acts as a competing endogenous RNA (ceRNA) [158,159], by depleting microRNAs. As a consequence, specific RNA targets cannot be degraded, which may lead to the dysregulation of downstream genes [160,161]. So far, both epigenetic and genetic changes have been observed in relation to the Xi of cancer cells, but whether these alterations are driving events that give a selective advantage to cancer cells is under debate. Nevertheless, evidence suggests that the Xi epigenetic status and XIST expression levels are potential cancer biomarkers as a readout for genomic instability or epigenomic changes. Therefore, understanding the factors and mechanisms that render and maintain the X chromosome inactive, both during embryonic development and in somatic cells during the maintenance phase of XCI, is of crucial importance.
3. Chromatin Modifiers That Act in XCI
The regulation of the X chromosome is controlled by chromatin modifiers that build up heterochromatin formation by deacetylating and methylating histone tails, finally leading to the DNA methylation of regulatory CpG islands (see Figure 2).
Specific enzymes that play a central role in XCI are HDACs, the PRC1 and PRC2 complexes, and DNMTs (see Table 1). Recently, the SHARP protein has been identified as a direct Xist interactor. This protein bridges Xist to HDACs allowing for histone deacetylation at the X chromosome. This section discusses the current knowledge about SHARP and other key chromatin modifiers that are involved in XCI.
3.1. SHARP
SHARP is a protein of more than 400 kDa that contains four RRMs and a highly conserved C-terminal domain, called SPOC (Spen paralog and ortholog C-terminal domain), which is responsible for mediating the repressive function of SHARP [162]. There is a family of SPOC domain-containing proteins that includes RNA binding motif protein 15/one-twenty-two (RBM15/OTT1) and RNA binding motif protein 15B/one-twenty-two protein 3 (RBM15B/OTT3) [163,164,165], which have recently been linked to XCI [52,166,167].
The SHARP-encoding gene was originally identified by Newberry and colleagues while screening an expression library from mouse brain to identify novel interaction partners for the Homeo domain transcriptional repressor Msx2 (Homolog of Muscle Segment Homeobox 2, Msh Homeobox 2) using a Farwestern approach. They named the interacting protein MINT. The full length protein was reported to have 3576 amino acids and three RRMs within the amino-terminal part [168]. Because 68 missing residues in the amino-terminal part of the original MINT protein analysis, they did not identify RRM1 in this report [168]. Subsequently, Shi and colleagues performed a yeast two hybrid screen using a mouse whole embryo E17 library and the carboxy-terminus of NCoR2 (also known as SMRT). Sequence information from the mouse clone was used to screen a human cDNA library. The full length human cDNA coded for a 3651 amino acid protein, which they named SHARP. The SMRT interacting protein fragment that was identified by Shi et al. corresponded to the C-terminus of SHARP, which they named Repression Domain (RD, now referred to as SPOC) [50]. Interestingly, the cDNA clone encoding for the MSX2-interacting protein fragment that was isolated by Newberry et al. [168] corresponds to amino acids 2138 to 2462 in MINT (Q62504.2), and it is closely related to the later reported receptor interaction domain (RID) in the human MINT homolog SHARP [50]. To search for novel components of the Notch signaling pathway, we also performed a yeast two hybrid screen using a human embryonic brain library and the DNA binding transcription factor (TF) of Notch signaling, recognition signal binding protein for immunoglobulin kappa J region [RBPJ; also known as CSL (CBF1, Suppressor of Hairless, Lag-1)], as a bait. This screen identified a cDNA encoding for a protein identical to SHARP. SHARP (NP_055816.2) consists of 3664 residues and the RRM1 was identified at the very amino-terminus [169]. The highly conserved RBPJ interaction domain (RBPID) of SHARP was fine mapped from residues 2882 (2776 in MINT) to 2839 (2814 in MINT) and structure information of the RBP-J-SHARP/MINT complex became available meanwhile [170]. Interestingly, BLAST analysis identifies 79% identity between the human and mouse SHARP proteins.
In regard to the in vivo function of SHARP, its mouse homolog, MINT, has been studied, making use of knockout models. Mint knockouts were primarily analyzed for its function in the Notch signal transduction pathway, since SHARP is a pivotal cofactor at Notch target genes [162,169,170,171,172,173,174,175]. Mint knockout mice are embryonic lethal at around day E12.5-14.5 and they show, amongst others, cardiac and pancreatic defects, as well as an increased number of marginal zone B cells [176]. Whether there is a difference in lethality between male and female embryos was not studied. Further studies making use of Mint knockout mice unveiled the role of Mint in the thymus supporting early T-cell development [177]. Additional studies have described the function of MINT, in regulating the expression of the osteocalcin-encoding gene [168,178] and collagen type II alpha 1 chain (Col2a1 [179]).
Early studies described SHARP as a regulator of nuclear receptors-dependent transcription by recruiting the HDACs-containing corepressor SMRT complex via the SPOC domain [50]. The same study also highlighted the ability of SHARP to bind lncRNAs, such as steroid receptor coactivator (SRA), to modulate gene expression [50]. However, SHARP is not an exclusive regulator of nuclear receptors-dependent transcription. In fact, it has also been linked to the highly conserved Notch signaling pathway that is involved in regulating different developmental and differentiation events and it is frequently dysregulated in cancer ([180,181,182], see Table 1). As discussed above, SHARP was recently identified in a screen for Xist RNA binding proteins (see Table 1) and it was shown to be essential for X-inactivation in embryonic stem cells (ESCs) [51,52,53,54,55,129]. However, SHARP does not bind exclusively SRA and Xist but also retroviral RNAs that are characterized by regions with structural similarity to the A-repeat of Xist [128], which is required for the Xist/SHARP interaction [51,53,130]. SHARP was shown to bind to the SRA lncRNA by its RRMs. The four RRMs of SHARP are located at its amino-terminal portion (aa 1–600, see Figure 3A). Solving the crystal structure of SHARP RRM2, RRM3, and RRM4 (see Figure 3B), Arieti and colleagues could demonstrate that RRM3 and RRM4 form an inter-domain platform (see Figure 3B orange and red), whereas RRM2 is not part of this platform (see Figure 3B, yellow). Additional RNA binding studies showed that the RRM3/RRM4 platform interacts with the H12-H13 region of SRA, whereas RRM2 seems not to be involved in this interaction [183]. Moreover, the role of RRM1 (see Figure 3A) in RNA binding has not been elucidated. Structure homology modeling suggests that the highly conserved amino acids 6 to 81 in SHARP form a typical RRM topology (see Figure 3C). In addition, structure alignments of RRMs identify the typical amino acids in essential positions needed for interactions with nucleotides (see Figure 3D–F). Although RRM2 alone seems not to be involved in SRA binding, one could speculate that RRM1 and RRM2 also form an intermolecular platform to bind specific lncRNAs. Structure analyses of a SHARP protein, including all four RRMs together with additional RNA binding studies, will give more insights into the exact role of each RRM and potential cooperative effects of RNA binding and regulation of gene transcription. In summary, lncRNA-mediated gene regulation by SHARP needs at least the amino terminal RRMs for RNA binding as well as the carboxy-terminal SPOC domain (see Figure 3A) for the recruitment of epigenetic modifiers.
In this review, we summarize and extract what is known regarding SHARP as a transcription and chromatin regulator, which will be useful for understanding its recently emerged role in XCI.
3.1.1. SHARP in Chromatin Regulation
SHARP has been characterized as the central corepressor at Notch target genes, forming the bridge between the transcription factor RBPJ and several corepressors [181]. In short, RBPJ either interacts with the Notch coactivator or with the SHARP corepressor based on the activation status of the Notch signaling pathway. This repressor-activator switch is carefully controlled by the Notch intracellular domain (NICD) and, consequently, its turnover determines the amplitude and duration of the Notch response [162,180,181,185].
SHARP has been initially characterized as a key component of the RBPJ-associated corepressor complex [169] by functioning as a platform for the recruitment of additional corepressors, such as eight-twenty-one/myeloid translocational gene 8 (ETO/MTG8) and C-terminal binding protein/CtBP-interacting protein (CtBP/CtIP), finally bridging RBPJ to HDACs [172,173,174]. Recent studies have further defined the interactome of the SPOC domain of SHARP (referred to as SPOCome) and shed light on a surprising and unexpected mechanism. In fact, SHARP does not exclusively interact with corepressors via its SPOC domain, but also with coactivators [162,171]. In this study, we observed that SPOC is able to interact in a mutually exclusive fashion with the corepressor NCoR, an ortholog of SMRT (also known as NCoR2) that has been previously linked to the Notch signaling pathway similarly to SMRT [186,187,188,189,190], or with the coactivator lysine (K) methyltransferase 2D (KMT2D) [171]. Given that the KMT2D complex has histone H3K4 methyltransferase activity and that the NCoR complex contains HDACs, this competition allows for balancing acetylation on lysine 27 of histone H3 (H3K27ac) and trimethylation of H3K4 (H3K4me3), fine-tuning the expression of Notch target genes [171]. It must be noted that this NCoR-KMT2D competition is strongly dependent on the phosphorylation status of NcoR, which has been previously shown to be phosphorylated on two highly conserved serine residues within its C-terminal LSDSD motif [191,192,193,194,195]. We observed that phospho-NCoR outcompetes KMT2D for binding to SPOC promoting the repression of Notch target genes [162,171]. In conclusion, this study emphasized that SHARP is more than a simple corepressor, but it operates as a poising factor that balances the repression and activation of Notch target genes. Therefore, SHARP might be a potential therapeutic target for both cancers in which Notch has been defined as a tumor suppressor as well as malignancies in which Notch acts as an oncogene. To reach this goal, thermodynamic studies [196] and the recently characterized crystal structure of the RBPJ/SHARP complex [170] may be useful. In fact, these data may allow developing small molecules modulating the RBPJ/SHARP interaction. These molecules can be further validated in biochemical and functional analysis with the final goal to get clinical relevance as therapeutic drugs. Additionally, it might be possible to develop inhibitors of the SPOC-NCoR/SMRT interaction to block the repressive activity of SHARP and reactivating the Notch pathway in those tumors in which Notch acts as a tumor suppressor. Additionally, in this case, the crystal structure of the SPOC-NCoR/SMRT complex may be instructive [197].
As discussed above, SHARP promotes XCI via the recruitment of deacetylating complexes through its SPOC domain [51,52,53,54,55,129]. It is important to also note that RBM15/OTT1 has been identified as a Xist interactor, marking further the importance of SPOC domain-containing proteins in XCI [52,54]. RBM15/OTT1 and its paralog RBM15B/OTT3 bridge Xist to the m6A methylation machinery promoting m6A methylation of Xist [167]. In line with that, Wilms tumor 1 (WT1)-associated (WTAP) and VIRILIZER proteins, subunits of the m6A methylation machinery have been previously identified as Xist interactors [52] and the Drosophila homolog of RBM15/OTT1, known as Nito, is also involved in m6A RNA methylation [198].
In addition, the SPOC domain of SHARP has been described to interact with the ubiquitin-conjugating enzyme (E2) UbcH8 (also known as UBE2L6 (ubiquitin conjugating enzyme E2 L6)) decreasing Notch-dependent promoter activity in a SHARP-dependent fashion [199]. It has also been suggested that SHARP homodimerizes through its SPOC domain, leading to a reduction of its repressive activity through RBPJ in luciferase assays [200]. Once more, these studies highlight the complex function of SHARP marking the requirement for a better comprehension of its molecular functions.
3.1.2. SHARP Directly Interacts with the NCoR/HDAC Complex
As mentioned above, SHARP was initially identified to interact in a yeast-2-hybrid screen with SMRT [50]. Later on we could show that it interacts with NCoR in a phospho-dependent manner [171]. The NcoR and SMRT complexes are composed of several subunits with different specific functions (see Table 1) and mutations of their components have been observed in cancer, as well as intellectual and developmental disorders (see Table 1). Purification studies of the NCoR and SMRT complexes unveiled their subunits composition identifying the following ones [79,80,201,202,203,204,205]: SMRT or NCoR1 themself; transducin β-like protein 1 (TBL1) whose gene is located on the X chromosome and mutated in human sensorineural deafness [206]; transducing β-like 1 (TBL1)-related protein (TBLR1); the deacetylase HDAC3; and, G-protein pathway suppressor 2 (GPS2), an intracellular signaling protein. Structural studies have also shed light on the interaction between HDAC3 and SMRT [207]. Smrt knockout results in severe heart defects that lead to the death of most of the embryos at day E16.5 [208]. Jepsen and colleagues could overcome this limit using myocyte-specific reexpression of Smrt and observed that Smrt is also required for forebrain development [190]. On the other hand, Ncor knockout leads to defects in erythropoiesis, T cell, and neural development [209]. Finally, Hdac3 knockout is embryonic lethal and its conditional knockout in liver results in hepatocellular carcinoma [210,211].
Among all of the subunits of the NCoR and SMRT complexes the HDACs that confer deacetylation activity to those complexes and determine their transcriptional repression ability are probably the most important subunits. However, recent studies have also suggested that HDACs can play a positive role in transcription and this is also true for HDAC3 [212,213,214]. This positive function is given by the fact that HDACs deacetylate histone proteins, but also non histone proteins. We have found that HDAC3 promotes the deacetylation of NICD1 increasing its stability and as consequence its transcriptional activity [212]. It is proposed that HDAC3 is found exclusively within the NCoR and SMRT complexes and required for their catalytic activity [215,216]. However, it still needs to be investigated whether HDAC3 can also work independently of the NCoR and SMRT complexes to fulfill its positive role in gene transcription. In fact, it cannot be excluded that other proteins that are different than SMRT and NCoR may be able to stimulate the catalytic activity of HDAC3.
NCoR and SMRT have been linked to the regulation of several different DNA binding proteins, such as the transcriptional repressor B cell lymphoma 6 (BCL6) [217,218], thyroid hormone receptor (THR) [203], RBPJ [171,186,187,188,189], the oncogenic fusion protein acute myeloid leukemia 1/eight-twenty-one (AML1/ETO) [219], TEL and c-JUN [220], and REV-ERBα, which is a transcription factor that is involved in the circadian clock [221]. SHARP interacts with NCoR and SMRT [171,222], recruiting them to the DNA, as we have briefly described above. Structural studies clarified how this interaction occurs and how it is regulated: it involves arginine (R) 3552 and 3554 of SHARP (within the SPOC domain) and serines (S) 2449 and 2451 of NCoR [162,171,223]. Furthermore, this interaction is dependent on the phosphorylation status of these serine residues within NCoR and at least one of them is phosphorylated by casein kinase 2 (CK2) [171,191,192,193,194,195]. Again, the availability of the crystal structure of the SPOC domain of SHARP in combination with phosphoSMRT [224] may be useful in developing molecules to inhibit this interaction as a potential therapeutic option. In addition, the inhibitors of CK2 may be a powerful tool to prevent the interaction between SHARP and SMRT or NCoR. It is important to note that SHARP is phosphorylated by p21-activated kinase 1 (PAK1) within the SPOC domain at S3486 and at threonine (T) 3568 [225]. PAK1-dependent phosphorylation of SHARP augments its repressive activity in luciferase assays [225]. However, whether this phosphorylation impacts on the SHARP-SMRT/NCoR interaction is unknown, and it would be important to evaluate that with inhibitors of PAK1 to potentially destabilize this interaction.
3.1.3. Pathological Deregulation of SHARP
SHARP has been linked to several diseases both because of mutations that occur within the gene or because of its altered function, localization, and/or expression (see Table 1). Frameshift and non-sense mutations of the SPEN gene have been described in adenoid cystic carcinoma [131] and in mantle cell lymphoma (MCL) [132,133,134]. SHARP mutations have been also described in diffuse large B-cell lymphoma (DLBCL) [135], in splenic marginal zone lymphoma (SMZL) [136,137], in pancreatic adenosquamous carcinoma (PASC) [138], as well as in neurodevelopmental disorders (NDDs) [139]. However, whether these mutations deregulate the Notch pathway and/or XCI is not clear. The identification of mutations in other Notch pathway components in the same type of tumor [131,132,133,134,136,137] would suggest that these mutations may have a negative impact on the Notch signaling pathway. However, SHARP does not exclusively regulate XCI and the Notch signaling pathway; in fact, it regulates the estrogen receptor α (ERα)-dependent transcription and mutations of SHARP have been detected in breast cancer, where it acts as a tumor suppressor [226] Similarly, SHARP expression is upregulated in colorectal adenocarcinoma, where it is described to deregulate the Wnt signaling pathway [227].
SHARP is mislocalized in myotonic dystrophy [228], while, in acute myeloid leukemia (AML), it has been proposed to have an altered function as consequence of its interaction with the oncofusion protein AML1/ETO deregulating the Notch signaling pathway [173,174]. Following the same line of reasoning, mutations of the genes encoding for SHARP interactors might have a deleterious ending. This might be the case of subunits of the KMT2D complex; in fact, KMT2D and lysine demethylase 6A/ubiquitously transcribed tetratricopeptide repeat protein X-linked (KDM6A/UTX) mutations have been observed in patients that are affected by Kabuki Syndrome [229,230,231,232,233,234,235,236,237] and, in line with that, kmt2d knockout in zebrafish recapitulates the Kabuki phenotype and it is characterized by the deregulation of the Notch pathway [238].
Similarly to SHARP, the SPOC domain-containing RBM15/OTT1 protein has also been linked to diseases. For example, it is translocated in acute megakaryocytic leukemia [239,240] and significantly upregulated in patients with blast-crisis chronic myelogenous leukemia (CML) [241].
These studies further mark the relevance of the proteins containing a SPOC domain and the importance to better characterize the function of the SPOC domain in normal as well as pathological conditions. Hitherto, it remains completely unclear as to whether XCI is affected in the same diseases in which SHARP is dysfunctional.
3.2. PRC1 and PRC2
It has been shown that the lncRNA Xist is key for the recruitment of Polycomb complexes to the future Xi [65,77]. The Polycomb group (PcG) genes were originally identified in Drosophila melanogaster [242]. Their products are organized in two different multisubunit complexes that are known as PRC1 and PRC2, which are involved in building up a repressive chromatin environment. As previously introduced, PRC1 promotes H2AK119ub1, while PRC2 deposits H3K27me3. Recent studies also elucidated additional PRC complexes that differ from each other based on subunits composition [76].
All of the PRC1 complexes contain RING1A or 1B (see Table 1). The subcomplex type is defined by the PcG ring finger protein (PCGF 1–6). RING and PCGF form a core unit that is common to all the PRC1 complexes, and this unit is associated with additional specific subunits, including polyhomeotic homolog (PHC, isoform 1–3), sex comb on midleg homolog (SCMH, isoform L1 or L2), and one of the chromobox homolog (CBX, either isoform 2, 4, 6, 7, or 8) proteins. Non-canonical PRC1 complexes do not contain CBX, but they still associate with RING1 and YY1 binding protein (RYBP and YAF2) cofactors. In the case of PRC2 complexes, EZH1/2, retinoblastoma binding protein (RBBP, it can be isoform 4 or 7), suppressor of zeste 12 (SUZ12), and embryonic ectoderm development (EED) form the core that associates with different subunits, giving rise to the PRC2.1 or PRC2.2 complexes. To note subunits of both PRC1, PRC2 as well as ncPRC1 have been observed in different types of cancer (see Table 1).
Polycomb complexes follow a specific sequence of events to help silence the future Xi (see Figure 2). First, non-canonical PRC1 is recruited via heterogeneous nuclear ribonucleoprotein K (hnRNPK), which also interacts with Xist [65,77]. This complex promotes H2AK119ub1 on the Xi in response to Xist expression, and this modification is recognized by the PRC2 complex through its subunit JARID2 [106]. PCR2 establishes H3K27me3 domains due to the activity of EZH1/2 [77]. Subsequently, H3K27me3 is recognized by the canonical PRC1, which enforces the silencing on the X chromosome [144,243]. Other lncRNAs might use similar mechanisms to promote the PRC2-dependent spreading of H3K27me3, for example, Airn and Kcnq1ot1 [244].
3.3. DNA Methyltransferases (DNMTs)
XCI is locked in through DNA methylation that occurs on the fifth carbon of cytosine, indicated as 5mC. 5mC usually occurs at regions of the genome that contain the CpG dinucleotide and it is enriched at repetitive sequences as well as within gene bodies. CpG dense regions are usually located near the Transcription Starting Site (TSS) of genes and they are defined as CpG islands or CGI [245]. Usually, the methylation of CGI is associated with gene silencing and it is highly stable. However, recent studies unveiled that hydroxylation of 5mC by ten-eleven translocation (TET) enzymes facilitates the rapid reactivation of silenced target genes [245].
DNA methylation is catalyzed by DNMTs, which are grouped into two main classes: the de novo DNMTs such as DNMT3A and DNMT3B establish the DNA methylation pattern during early embryogenesis, while the maintenance DNMT, DNMT1, restore the DNA methylation pattern after DNA replication [246,247,248]. 5mC marks are subsequently read by proteins that contain dedicated domains and that bridge 5mC to additional enzymes to further support the establishment of repressive chromatin [249]. Three different classes of 5mC readers are known: readers that contain the methyl binding domain (MBD); Kaiso and Kaiso-like proteins that contains the broad complex, tramtrack, and bric a brac/Pox virus and zinc finger (BTB/POZ) domain and Krüppel-like C2H2 zinc fingers; finally, proteins containing the SET and RING finger-associated (SRA) domain [250]. It is also important to note that unmethylated CGI are targets of dedicated proteins, for example, CXXC finger protein 1 (CFP1) [251,252].
The long term maintenance of X chromosome inactivation is achieved via DNA methylation that occurs at promoter-associated CGIs. This methylation is catalyzed by DNMT3B (see Figure 2 and Table 1), and it occurs with two different kinetics at different regions of the X chromosome [56]. At most CGIs, DNA methylation occurs slowly and requires the chromosomal binding of structural maintenance of chromosomes hinge domain-containing 1 (SMCHD1), while, at a small proportion of CGIs, DNA methylation is SMCHD1-independent through a very fast process.
4. Conclusions
LncRNAs are key players in many different cellular processes. Here, we have reviewed the recent discoveries how heterochromatin formation, initiated by lncRNA Xist, is established. XCI is a tightly regulated process that is controlled by multiple epigenetic regulators, amongst them SHARP, that, in recent years, has been shown to play a central role in the silencing of the future Xi. Mechanistically, it will be interesting to find out whether SHARP’s main function is to recruit the HDAC3-containing NCoR1/2 complexes or whether additional interaction partners will be required for XCI. Furthermore, the function of the N-terminal RRM1 of SHARP, most likely acting in concert with RRM2, remains to be elucidated. Alterations of SHARP and additional XCI-contributing factors have been identified in several cancer types, but to which extend they contribute to cancer progression remains to be determined, as well as whether these alterations affect the Xi status. Studies focusing on male and female specific differences could help to understand their potential therapeutic value.
Funding
B.D.G. is supported by a research grant of the University Medical Center Giessen and Marburg (UKGM) and by a Prize of the Justus Liebig University Giessen to B.D.G. The work was further supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—TRR81- A12 and BO 1639/9-1 to TB, SFB 1074/A03, OS 287/4-1 and GRK 2254/C4 to FO and the Von Behring-Röntgen foundation and Excellence Cluster for Cardio Pulmonary System (ECCPS) in Giessen to T.B.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gao, N.; Li, Y.; Li, J.; Gao, Z.; Yang, Z.; Li, Y.; Liu, H.; Fan, T. Long Non-Coding RNAs: The Regulatory Mechanisms, Research Strategies, and Future Directions in Cancers. Front. Oncol. 2020, 10, 598817. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Thum, T.; Condorelli, G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ. Res. 2015, 116, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- Liu, F.; Somarowthu, S.; Pyle, A.M. Visualizing the secondary and tertiary architectural domains of lncRNA RepA. Nat. Chem. Biol. 2017, 13, 282–289. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, Z.; Shi, H.; Li, H.; Li, L.; Fang, R.; Cai, X.; Liu, B.; Zhang, X.; Ye, L. HBXIP and LSD1 Scaffolded by lncRNA Hotair Mediate Transcriptional Activation by c-Myc. Cancer Res. 2016, 76, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.M.; Yang, F.Q.; Chen, S.J.; Che, J.; Zheng, J.H. Upregulation of long non-coding RNA MALAT1 correlates with tumor progression and poor prognosis in clear cell renal cell carcinoma. Tumour Biol. 2015, 36, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.X.; Wang, H.J.; Li, X.R.; Li, T.; Su, G.; Yang, P.; Wu, J.W. Long noncoding RNA MALAT1 associates with the malignant status and poor prognosis in glioma. Tumour Biol. 2015, 36, 3355–3359. [Google Scholar] [CrossRef]
- Dong, Y.; Liang, G.; Yuan, B.; Yang, C.; Gao, R.; Zhou, X. MALAT1 promotes the proliferation and metastasis of osteosarcoma cells by activating the PI3K/Akt pathway. Tumour Biol. 2015, 36, 1477–1486. [Google Scholar] [CrossRef]
- Cho, S.F.; Chang, Y.C.; Chang, C.S.; Lin, S.F.; Liu, Y.C.; Hsiao, H.H.; Chang, J.G.; Liu, T.C. MALAT1 long non-coding RNA is overexpressed in multiple myeloma and may serve as a marker to predict disease progression. BMC Cancer 2014, 14, 809. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.T.; Shi, D.B.; Wang, Y.W.; Li, X.X.; Xu, Y.; Tripathi, P.; Gu, W.L.; Cai, G.X.; Cai, S.J. High expression of lncRNA MALAT1 suggests a biomarker of poor prognosis in colorectal cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 3174–3181. [Google Scholar]
- Hirata, H.; Hinoda, Y.; Shahryari, V.; Deng, G.; Nakajima, K.; Tabatabai, Z.L.; Ishii, N.; Dahiya, R. Long Noncoding RNA MALAT1 Promotes Aggressive Renal Cell Carcinoma through Ezh2 and Interacts with miR-205. Cancer Res. 2015, 75, 1322–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.S.; Wang, X.A.; Wu, W.G.; Hu, Y.P.; Li, M.L.; Ding, Q.; Weng, H.; Shu, Y.J.; Liu, T.Y.; Jiang, L.; et al. MALAT1 promotes the proliferation and metastasis of gallbladder cancer cells by activating the ERK/MAPK pathway. Cancer Biol. Ther. 2014, 15, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Su, L.; Chen, X.; Li, P.; Cai, Q.; Yu, B.; Liu, B.; Wu, W.; Zhu, Z. MALAT1 promotes cell proliferation in gastric cancer by recruiting SF2/ASF. Biomed. Pharmacother. 2014, 68, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xue, D.; Li, Y.; Pan, X.; Zhang, X.; Kuang, B.; Zhou, M.; Li, X.; Xiong, W.; Li, G.; et al. The Long Noncoding RNA MALAT-1 is A Novel Biomarker in Various Cancers: A Meta-analysis Based on the GEO Database and Literature. J. Cancer 2016, 7, 991–1001. [Google Scholar] [CrossRef]
- Poirier, F.; Chan, C.T.; Timmons, P.M.; Robertson, E.J.; Evans, M.J.; Rigby, P.W. The murine H19 gene is activated during embryonic stem cell differentiation in vitro and at the time of implantation in the developing embryo. Development 1991, 113, 1105–1114. [Google Scholar] [PubMed]
- Gabory, A.; Jammes, H.; Dandolo, L. The H19 locus: Role of an imprinted non-coding RNA in growth and development. Bioessays 2010, 32, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Gabory, A.; Ripoche, M.A.; Yoshimizu, T.; Dandolo, L. The H19 gene: Regulation and function of a non-coding RNA. Cytogenet. Genome Res. 2006, 113, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, B.; Li, J.; Su, L.; Yan, M.; Zhu, Z.; Liu, B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget 2014, 5, 2318–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Bi, J.; Xue, X.; Zheng, L.; Zhi, K.; Hua, J.; Fang, G. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 2012, 279, 3159–3165. [Google Scholar] [CrossRef]
- Yoshimura, H.; Matsuda, Y.; Yamamoto, M.; Kamiya, S.; Ishiwata, T. Expression and role of long non-coding RNA H19 in carcinogenesis. Front. Biosci. (Landmark Ed.) 2018, 23, 614–625. [Google Scholar] [CrossRef]
- Xie, S.S.; Jin, J.; Xu, X.; Zhuo, W.; Zhou, T.H. Emerging roles of non-coding RNAs in gastric cancer: Pathogenesis and clinical implications. World J. Gastroenterol. 2016, 22, 1213–1223. [Google Scholar] [CrossRef]
- Zhang, E.B.; Kong, R.; Yin, D.D.; You, L.H.; Sun, M.; Han, L.; Xu, T.P.; Xia, R.; Yang, J.S.; De, W.; et al. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget 2014, 5, 2276–2292. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Xue, X.; Zheng, L.; Bi, J.; Zhou, Y.; Zhi, K.; Gu, Y.; Fang, G. Long non-coding RNA GHET1 promotes gastric carcinoma cell proliferation by increasing c-Myc mRNA stability. FEBS J. 2014, 281, 802–813. [Google Scholar] [CrossRef]
- Kong, R.; Zhang, E.B.; Yin, D.D.; You, L.H.; Xu, T.P.; Chen, W.M.; Xia, R.; Wan, L.; Sun, M.; Wang, Z.X.; et al. Long noncoding RNA PVT1 indicates a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically regulating p15 and p16. Mol. Cancer 2015, 14, 82. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Wu, G.; Fan, H.; Wu, J.; Feng, J. Long noncoding RNA SPRY4-IT1 predicts poor patient prognosis and promotes tumorigenesis in gastric cancer. Tumour. Biol. 2015, 36, 6751–6758. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.J.; Xie, S.L.; Li, Q.; Ma, J.; Wang, G.Y. Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J. Int. Med. Res. 2011, 39, 2119–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.Y.; Yu, Q.M.; Du, Y.A.; Yang, L.T.; Dong, R.Z.; Huang, L.; Yu, P.F.; Cheng, X.D. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int. J. Biol. Sci. 2013, 9, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Jutooru, I.; Chadalapaka, G.; Johnson, G.; Frank, J.; Burghardt, R.; Kim, S.; Safe, S. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 2013, 32, 1616–1625. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, T.; Okamoto, K.; Sugihara, H.; Hattori, T.; Reeve, A.E.; Ogawa, O.; Okada, Y. The roles of supernumerical X chromosomes and XIST expression in testicular germ cell tumors. J. Urol 2003, 169, 1546–1552. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Silver, D.P.; Greenberg, R.A.; Avni, D.; Drapkin, R.; Miron, A.; Mok, S.C.; Randrianarison, V.; Brodie, S.; Salstrom, J.; et al. BRCA1 supports XIST RNA concentration on the inactive X chromosome. Cell 2002, 111, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Pageau, G.J.; Hall, L.L.; Lawrence, J.B. BRCA1 does not paint the inactive X to localize XIST RNA but may contribute to broad changes in cancer that impact XIST and Xi heterochromatin. J. Cell Biochem. 2007, 100, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.L.; Wang, Z.C.; de Nicolo, A.; Lu, X.; Brown, M.; Miron, A.; Liao, X.; Iglehart, J.D.; Livingston, D.M.; Ganesan, S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell 2006, 9, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.P.; Dimitrov, S.D.; Feunteun, J.; Gelman, R.; Drapkin, R.; Lu, S.D.; Shestakova, E.; Velmurugan, S.; Denunzio, N.; Dragomir, S.; et al. Further evidence for BRCA1 communication with the inactive X chromosome. Cell 2007, 128, 991–1002. [Google Scholar] [CrossRef] [Green Version]
- Sirchia, S.M.; Ramoscelli, L.; Grati, F.R.; Barbera, F.; Coradini, D.; Rossella, F.; Porta, G.; Lesma, E.; Ruggeri, A.; Radice, P.; et al. Loss of the inactive X chromosome and replication of the active X in BRCA1-defective and wild-type breast cancer cells. Cancer Res. 2005, 65, 2139–2146. [Google Scholar] [CrossRef] [Green Version]
- Sirchia, S.M.; Tabano, S.; Monti, L.; Recalcati, M.P.; Gariboldi, M.; Grati, F.R.; Porta, G.; Finelli, P.; Radice, P.; Miozzo, M. Misbehaviour of XIST RNA in breast cancer cells. PLoS ONE 2009, 4, e5559. [Google Scholar] [CrossRef]
- Vincent-Salomon, A.; Ganem-Elbaz, C.; Manie, E.; Raynal, V.; Sastre-Garau, X.; Stoppa-Lyonnet, D.; Stern, M.H.; Heard, E. X inactive-specific transcript RNA coating and genetic instability of the X chromosome in BRCA1 breast tumors. Cancer Res. 2007, 67, 5134–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benoit, M.H.; Hudson, T.J.; Maire, G.; Squire, J.A.; Arcand, S.L.; Provencher, D.; Mes-Masson, A.M.; Tonin, P.N. Global analysis of chromosome X gene expression in primary cultuRes. of normal ovarian surface epithelial cells and epithelial ovarian cancer cell lines. Int. J. Oncol. 2007, 30, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawakami, T.; Zhang, C.; Taniguchi, T.; Kim, C.J.; Okada, Y.; Sugihara, H.; Hattori, T.; Reeve, A.E.; Ogawa, O.; Okamoto, K. Characterization of loss-of-inactive X in Klinefelter syndrome and female-derived cancer cells. Oncogene 2004, 23, 6163–6169. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; del Rosario, B.C.; Szanto, A.; Ogawa, Y.; Jeon, Y.; Lee, J.T. Jpx RNA activates Xist by evicting CTCF. Cell 2013, 153, 1537–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barakat, T.S.; Loos, F.; van Staveren, S.; Myronova, E.; Ghazvini, M.; Grootegoed, J.A.; Gribnau, J. The trans-activator RNF12 and cis-acting elements effectuate X chromosome inactivation independent of X-pairing. Mol. Cell 2014, 53, 965–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furlan, G.; Gutierrez-Hernandez, N.; Huret, C.; Galupa, R.; van Bemmel, J.G.; Romito, A.; Heard, E.; Morey, C.; Rougeulle, C. The Ftx Noncoding Locus Controls X Chromosome Inactivation Independently of Its RNA Products. Mol. Cell 2018, 70, 462–472.e468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutzel, V.; Okamoto, I.; Dunkel, I.; Saitou, M.; Giorgetti, L.; Heard, E.; Schulz, E.G. A symmetric toggle switch explains the onset of random X inactivation in different mammals. Nat. Struct. Mol. Biol. 2019, 26, 350–360. [Google Scholar] [CrossRef]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat. Genet. 2002, 30, 167–174. [Google Scholar] [CrossRef]
- Shi, Y.; Downes, M.; Xie, W.; Kao, H.Y.; Ordentlich, P.; Tsai, C.C.; Hon, M.; Evans, R.M. Sharp, an inducible cofactor that integrates nuclear receptor repression and activation. Genes Dev. 2001, 15, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Moindrot, B.; Cerase, A.; Coker, H.; Masui, O.; Grijzenhout, A.; Pintacuda, G.; Schermelleh, L.; Nesterova, T.B.; Brockdorff, N. A Pooled shRNA Screen Identifies Rbm15, Spen, and Wtap as Factors Required for Xist RNA-Mediated Silencing. Cell Rep. 2015, 12, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Monfort, A.; di Minin, G.; Postlmayr, A.; Freimann, R.; Arieti, F.; Thore, S.; Wutz, A. Identification of Spen as a Crucial Factor for Xist Function through Forward Genetic Screening in Haploid Embryonic Stem Cells. Cell Rep. 2015, 12, 554–561. [Google Scholar] [CrossRef] [Green Version]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Dossin, F.; Pinheiro, I.; Zylicz, J.J.; Roensch, J.; Collombet, S.; le Saux, A.; Chelmicki, T.; Attia, M.; Kapoor, V.; Zhan, Y.; et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature 2020, 578, 455–460. [Google Scholar] [CrossRef]
- Gendrel, A.V.; Apedaile, A.; Coker, H.; Termanis, A.; Zvetkova, I.; Godwin, J.; Tang, Y.A.; Huntley, D.; Montana, G.; Taylor, S.; et al. Smchd1-dependent and -independent pathways determine developmental dynamics of CpG island methylation on the inactive X chromosome. Dev. Cell 2012, 23, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.L.; Rigolet, M.; Bourc’his, D.; Nigon, F.; Bokesoy, I.; Fryns, J.P.; Hulten, M.; Jonveaux, P.; Maraschio, P.; Megarbane, A.; et al. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum. Mutat. 2005, 25, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, E.; Lev, A.; Eyal, E.; Barel, O.; Kol, N.; Barhom, S.F.; Pode-Shakked, B.; Anikster, Y.; Somech, R.; Simon, A.J. A Novel Mutation in a Critical Region for the Methyl Donor Binding in DNMT3B Causes Immunodeficiency, Centromeric Instability, and Facial Anomalies Syndrome (ICF). J. Clin. Immunol. 2016, 36, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Shirohzu, H.; Kubota, T.; Kumazawa, A.; Sado, T.; Chijiwa, T.; Inagaki, K.; Suetake, I.; Tajima, S.; Wakui, K.; Miki, Y.; et al. Three novel DNMT3B mutations in Japanese patients with ICF syndrome. Am. J. Med. Genet. 2002, 112, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Pequignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjostrom, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef]
- Herold, T.; Metzeler, K.H.; Vosberg, S.; Hartmann, L.; Jurinovic, V.; Opatz, S.; Konstandin, N.P.; Schneider, S.; Zellmeier, E.; Ksienzyk, B.; et al. Acute myeloid leukemia with del(9q) is characterized by frequent mutations of NPM1, DNMT3A, WT1 and low expression of TLE4. Genes Chromosomes Cancer 2017, 56, 75–86. [Google Scholar] [CrossRef] [PubMed]
- van den Boogaard, M.L.; Lemmers, R.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Pintacuda, G.; Wei, G.; Roustan, C.; Kirmizitas, B.A.; Solcan, N.; Cerase, A.; Castello, A.; Mohammed, S.; Moindrot, B.; Nesterova, T.B.; et al. hnRNPK Recruits PCGF3/5-PRC1 to the Xist RNA B-Repeat to Establish Polycomb-Mediated Chromosomal Silencing. Mol. Cell 2017, 68, 955–969.e910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamoto, M.Y.; Lammer, N.C.; Batey, R.T.; Wuttke, D.S. hnRNPK recognition of the B motif of Xist and other biological RNAs. Nucleic Acids Res. 2020, 48, 9320–9335. [Google Scholar] [CrossRef]
- Gallardo, M.; Lee, H.J.; Zhang, X.; Bueso-Ramos, C.; Pageon, L.R.; McArthur, M.; Multani, A.; Nazha, A.; Manshouri, T.; Parker-Thornburg, J.; et al. hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies. Cancer Cell 2015, 28, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Au, P.Y.B.; Goedhart, C.; Ferguson, M.; Breckpot, J.; Devriendt, K.; Wierenga, K.; Fanning, E.; Grange, D.K.; Graham, G.E.; Galarreta, C.; et al. Phenotypic spectrum of Au-Kline syndrome: A report of six new cases and review of the literature. Eur. J. Hum. Genet. 2018, 26, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Bastidas-Torres, A.N.; Cats, D.; Mei, H.; Szuhai, K.; Willemze, R.; Vermeer, M.H.; Tensen, C.P. Genomic analysis reveals recurrent deletion of JAK-STAT signaling inhibitors HNRNPK and SOCS1 in mycosis fungoides. Genes Chromosomes Cancer 2018, 57, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, L.; Pagnamenta, A.T.; Lise, S.; Clasper, S.; Stewart, H.; Akha, E.S.; Quaghebeur, G.; Knight, S.J.; Keays, D.A.; Taylor, J.C.; et al. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin. Genet. 2016, 90, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Dentici, M.L.; Barresi, S.; Niceta, M.; Pantaleoni, F.; Pizzi, S.; Dallapiccola, B.; Tartaglia, M.; Digilio, M.C. Clinical spectrum of Kabuki-like syndrome caused by HNRNPK haploinsufficiency. Clin. Genet. 2018, 93, 401–407. [Google Scholar] [CrossRef]
- Miyake, N.; Inaba, M.; Mizuno, S.; Shiina, M.; Imagawa, E.; Miyatake, S.; Nakashima, M.; Mizuguchi, T.; Takata, A.; Ogata, K.; et al. A case of atypical Kabuki syndrome arising from a novel missense variant in HNRNPK. Clin. Genet. 2017, 92, 554–555. [Google Scholar] [CrossRef] [PubMed]
- Naarmann-de-Vries, I.S.; Sackmann, Y.; Klein, F.; Ostareck-Lederer, A.; Ostareck, D.H.; Jost, E.; Ehninger, G.; Brummendorf, T.H.; Marx, G.; Rollig, C.; et al. Characterization of acute myeloid leukemia with del(9q)—Impact of the genes in the minimally deleted region. Leuk Res. 2019, 76, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Maystadt, I.; Deprez, M.; Moortgat, S.; Benoit, V.; Karadurmus, D. A second case of Okamoto syndrome caused by HNRNPK mutation. Am. J. Med. Genet. A 2020, 182, 1537–1539. [Google Scholar] [CrossRef]
- Okamoto, N. Okamoto syndrome has featuRes. overlapping with Au-Kline syndrome and is caused by HNRNPK mutation. Am. J. Med. Genet. A 2019, 179, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, V.; Mocavini, I.; di Croce, L. Polycomb complexes in normal and malignant hematopoiesis. J. Cell Biol. 2019, 218, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.; Pintacuda, G.; Masui, O.; Koseki, Y.; Gdula, M.; Cerase, A.; Brown, D.; Mould, A.; Innocent, C.; Nakayama, M.; et al. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 2017, 356, 1081–1084. [Google Scholar] [CrossRef]
- Palau, A.; Garz, A.K.; Diesch, J.; Zwick, A.; Malinverni, R.; Valero, V.; Lappin, K.; Casquero, R.; Lennartsson, A.; Zuber, J.; et al. Polycomb protein RING1A limits hematopoietic differentiation in myelodysplastic syndromes. Oncotarget 2017, 8, 115002–115017. [Google Scholar] [CrossRef]
- Zhang, J.; Kalkum, M.; Chait, B.T.; Roeder, R.G. The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell 2002, 9, 611–623. [Google Scholar] [CrossRef]
- Underhill, C.; Qutob, M.S.; Yee, S.P.; Torchia, J. A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J. Biol. Chem. 2000, 275, 40463–40470. [Google Scholar] [CrossRef] [Green Version]
- Pareja, F.; Ferrando, L.; Lee, S.S.K.; Beca, F.; Selenica, P.; Brown, D.N.; Farmanbar, A.; da Cruz-Paula, A.; Vahdatinia, M.; Zhang, H.; et al. The genomic landscape of metastatic histologic special types of invasive breast cancer. NPJ Breast Cancer 2020, 6, 53. [Google Scholar] [CrossRef]
- Nishi, A.; Numata, S.; Tajima, A.; Zhu, X.; Ito, K.; Saito, A.; Kato, Y.; Kinoshita, M.; Shimodera, S.; Ono, S.; et al. De novo non-synonymous TBL1XR1 mutation alters Wnt signaling activity. Sci. Rep. 2017, 7, 2887. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Yoo, H.Y.; Lee, S.H.; Shin, S.; Kim, S.C.; Lee, S.; Joung, J.G.; Nam, J.Y.; Ryu, D.; Yun, J.W.; et al. The mutational landscape of ocular marginal zone lymphoma identifies frequent alterations in TNFAIP3 followed by mutations in TBL1XR1 and CREBBP. Oncotarget 2017, 8, 17038–17049. [Google Scholar] [CrossRef] [Green Version]
- Heinen, C.A.; Jongejan, A.; Watson, P.J.; Redeker, B.; Boelen, A.; Boudzovitch-Surovtseva, O.; Forzano, F.; Hordijk, R.; Kelley, R.; Olney, A.H.; et al. A specific mutation in TBL1XR1 causes Pierpont syndrome. J. Med. Genet. 2016, 53, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz-Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Pons, L.; Cordier, M.P.; Labalme, A.; Till, M.; Louvrier, C.; Schluth-Bolard, C.; Lesca, G.; Edery, P.; Sanlaville, D. A new syndrome of intellectual disability with dysmorphism due to TBL1XR1 deletion. Am. J. Med. Genet. A 2015, 167A, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Saitsu, H.; Tohyama, J.; Walsh, T.; Kato, M.; Kobayashi, Y.; Lee, M.; Tsurusaki, Y.; Miyake, N.; Goto, Y.; Nishino, I.; et al. A girl with West syndrome and autistic featuRes. harboring a de novo TBL1XR1 mutation. J. Hum. Genet. 2014, 59, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Riehmer, V.; Erger, F.; Herkenrath, P.; Seland, S.; Jackels, M.; Wiater, A.; Heller, R.; Beck, B.B.; Netzer, C. A heritable microduplication encompassing TBL1XR1 causes a genomic sister-disorder for the 3q26.32 microdeletion syndrome. Am. J. Med. Genet. A 2017, 173, 2132–2138. [Google Scholar] [CrossRef]
- Ivanov, I.; Lo, K.C.; Hawthorn, L.; Cowell, J.K.; Ionov, Y. Identifying candidate colon cancer tumor suppressor genes using inhibition of nonsense-mediated mRNA decay in colon cancer cells. Oncogene 2007, 26, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Sinha, R.; Hoadley, K.A.; Jacobsen, A.S.; Reva, B.; Perou, C.M.; Sander, C.; Schultz, N. The molecular diversity of Luminal A breast tumors. Breast Cancer Res. Treat 2013, 141, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.R.; Huang, J.; Xu, Z.G.; Qian, B.Z.; Zhu, Z.D.; Yan, Q.; Cai, T.; Zhang, X.; Xiao, H.S.; Qu, J.; et al. Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc. Natl. Acad. Sci. USA 2001, 98, 15089–15094. [Google Scholar] [CrossRef] [Green Version]
- Awad, S.; Al-Dosari, M.S.; Al-Yacoub, N.; Colak, D.; Salih, M.A.; Alkuraya, F.S.; Poizat, C. Mutation in PHC1 implicates chromatin remodeling in primary microcephaly pathogenesis. Hum. Mol. Genet. 2013, 22, 2200–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, A.M.; Akunowicz, J.D.; Reveles, X.T.; Patel, B.B.; Saria, E.A.; Gorlick, R.G.; Naylor, S.L.; Leach, R.J.; Hansen, M.F. PHC3, a component of the hPRC-H complex, associates with E2F6 during G0 and is lost in osteosarcoma tumors. Oncogene 2007, 26, 1714–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.Y.; Sun, S.X.; Zhu, S.X.; Bu, J. Identification of the Roles of Chromobox Family Members in Gastric Cancer: A Study Based on Multiple Datasets. Biomed. Res. Int. 2020, 2020, 5306509. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders, S. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef]
- Lee, J.H.; Zhao, X.M.; Yoon, I.; Lee, J.Y.; Kwon, N.H.; Wang, Y.Y.; Lee, K.M.; Lee, M.J.; Kim, J.; Moon, H.G.; et al. Integrative analysis of mutational and transcriptional profiles reveals driver mutations of metastatic breast cancers. Cell Discov. 2016, 2, 16025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, I.; Batty, E.M.; Pole, J.C.; Blood, K.A.; Mo, S.; Cooke, S.L.; Ng, C.; Howe, K.L.; Chin, S.F.; Brenton, J.D.; et al. Structural analysis of the genome of breast cancer cell line ZR-75-30 identifies twelve expressed fusion genes. BMC Genom. 2012, 13, 719. [Google Scholar] [CrossRef] [Green Version]
- Turnpenny, P.D.; Wright, M.J.; Sloman, M.; Caswell, R.; van Essen, A.J.; Gerkes, E.; Pfundt, R.; White, S.M.; Shaul-Lotan, N.; Carpenter, L.; et al. Missense Mutations of the Pro65 Residue of PCGF2 Cause a Recognizable Syndrome Associated with Craniofacial, Neurological, Cardiovascular, and Skeletal Features. Am. J. Hum. Genet. 2018, 103, 786–793. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, Y.; Cheng, C.; Cui, H.; Cheng, L.; Kong, P.; Wang, J.; Li, Y.; Chen, W.; Song, B.; et al. Genomic analyses reveal mutational signatuRes. and frequently altered genes in esophageal squamous cell carcinoma. Am. J. Hum. Genet. 2015, 96, 597–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biason-Lauber, A.; Konrad, D.; Meyer, M.; DeBeaufort, C.; Schoenle, E.J. Ovaries and female phenotype in a girl with 46,XY karyotype and mutations in the CBX2 gene. Am. J. Hum. Genet. 2009, 84, 658–663. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, B.I.; Garcia, J.F.; Suela, J.; Mollejo, M.; Camacho, F.I.; Carro, A.; Montes, S.; Piris, M.A.; Cigudosa, J.C. Comparative genome profiling across subtypes of low-grade B-cell lymphoma identifies type-specific and common aberrations that target genes with a role in B-cell neoplasia. Haematologica 2008, 93, 670–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Rocha, S.T.; Boeva, V.; Escamilla-Del-Arenal, M.; Ancelin, K.; Granier, C.; Matias, N.R.; Sanulli, S.; Chow, J.; Schulz, E.; Picard, C.; et al. Jarid2 Is Implicated in the Initial Xist-Induced Targeting of PRC2 to the Inactive X Chromosome. Mol. Cell 2014, 53, 301–316. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Toki, T.; Okuno, Y.; Kanezaki, R.; Shiraishi, Y.; Sato-Otsubo, A.; Sanada, M.; Park, M.J.; Terui, K.; Suzuki, H.; et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. 2013, 45, 1293–1299. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [Green Version]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Ernst, T.; Chase, A.J.; Score, J.; Hidalgo-Curtis, C.E.; Bryant, C.; Jones, A.V.; Waghorn, K.; Zoi, K.; Ross, F.M.; Reiter, A.; et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 2010, 42, 722–726. [Google Scholar] [CrossRef]
- Nikoloski, G.; Langemeijer, S.M.; Kuiper, R.P.; Knops, R.; Massop, M.; Tonnissen, E.R.; van der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; de Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Bodor, C.; Grossmann, V.; Popov, N.; Okosun, J.; O’Riain, C.; Tan, K.; Marzec, J.; Araf, S.; Wang, J.; Lee, A.M.; et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood 2013, 122, 3165–3168. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Velusamy, T.; Rolland, D.; Sahasrabuddhe, A.A.; Chung, F.; Bailey, N.G.; Schrader, A.; Li, B.; Li, J.Z.; Ozel, A.B.; et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood 2014, 124, 1460–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Seki, M.; Kimura, S.; Isobe, T.; Yoshida, K.; Ueno, H.; Nakajima-Takagi, Y.; Wang, C.; Lin, L.; Kon, A.; Suzuki, H.; et al. Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntziachristos, P.; Tsirigos, A.; van Vlierberghe, P.; Nedjic, J.; Trimarchi, T.; Flaherty, M.S.; Ferres-Marco, D.; da Ros, V.; Tang, Z.; Siegle, J.; et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 2012, 18, 298–301. [Google Scholar] [CrossRef]
- Score, J.; Hidalgo-Curtis, C.; Jones, A.V.; Winkelmann, N.; Skinner, A.; Ward, D.; Zoi, K.; Ernst, T.; Stegelmann, F.; Dohner, K.; et al. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood 2012, 119, 1208–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, S.; Takenobu, H.; Kageyama, H.; Koseki, H.; Ishii, T.; Nakazawa, A.; Tatezaki, S.; Nakagawara, A.; Kamijo, T. Polycomb group molecule PHC3 regulates polycomb complex composition and prognosis of osteosarcoma. Cancer Sci. 2010, 101, 1646–1652. [Google Scholar] [CrossRef]
- Brecqueville, M.; Cervera, N.; Adelaide, J.; Rey, J.; Carbuccia, N.; Chaffanet, M.; Mozziconacci, M.J.; Vey, N.; Birnbaum, D.; Gelsi-Boyer, V.; et al. Mutations and deletions of the SUZ12 polycomb gene in myeloproliferative neoplasms. Blood Cancer J. 2011, 1, e33. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.B.; Sun, S.L.; Zheng, Q.L.; Zhang, L.; Zhu, Y.; Jin, G.H.; Xue, L.X. Genetic alteration and misexpression of Polycomb group genes in hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 2969–2979. [Google Scholar]
- Carter, A.C.; Xu, J.; Nakamoto, M.Y.; Wei, Y.; Zarnegar, B.J.; Shi, Q.; Broughton, J.P.; Ransom, R.C.; Salhotra, A.; Nagaraja, S.D.; et al. Spen links RNA-mediated endogenous retrovirus silencing and X chromosome inactivation. Elife 2020, 9. [Google Scholar] [CrossRef]
- Minajigi, A.; Froberg, J.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhali, M.; Haas, W.; et al. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotman, J.B.; Lee, D.M.; Cherney, R.E.; Kim, S.O.; Inoue, K.; Schertzer, M.D.; Bischoff, S.R.; Cowley, D.O.; Calabrese, J.M. Elements at the 5’ end of Xist harbor SPEN-independent transcriptional antiterminator activity. Nucleic Acids Res. 2020, 48, 10500–10517. [Google Scholar] [CrossRef]
- Stephens, P.J.; Davies, H.R.; Mitani, Y.; van Loo, P.; Shlien, A.; Tarpey, P.S.; Papaemmanuil, E.; Cheverton, A.; Bignell, G.R.; Butler, A.P.; et al. Whole exome sequencing of adenoid cystic carcinoma. J. Clin. Investig. 2013, 123, 2965–2968. [Google Scholar] [CrossRef]
- Hansen, M.H.; Cedile, O.; Blum, M.K.; Hansen, S.V.; Ebbesen, L.H.; Bentzen, H.H.N.; Thomassen, M.; Kruse, T.A.; Kavan, S.; Kjeldsen, E.; et al. Molecular characterization of sorted malignant B cells from patients clinically identified with mantle cell lymphoma. Exp. Hematol. 2020, 84, 7–18.e12. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Zhang, S.; Kanagal-Shamanna, R.; Ok, C.Y.; Nomie, K.; Gonzalez, G.N.; Gonzalez-Pagan, O.; Hill, H.A.; Lee, H.J.; Fayad, L.; et al. Genomic profiles and clinical outcomes of de novo blastoid/pleomorphic MCL are distinct from those of transformed MCL. Blood Adv. 2020, 4, 1038–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, H.A.; Qi, X.; Jain, P.; Nomie, K.; Wang, Y.; Zhou, S.; Wang, M.L. Genetic mutations and featuRes. of mantle cell lymphoma: A systematic review and meta-analysis. Blood Adv. 2020, 4, 2927–2938. [Google Scholar] [CrossRef]
- Hartert, K.T.; Wenzl, K.; Krull, J.E.; Manske, M.; Sarangi, V.; Asmann, Y.; Larson, M.C.; Maurer, M.J.; Slager, S.; Macon, W.R.; et al. Targeting of inflammatory pathways with R2CHOP in high-risk DLBCL. Leukemia 2020. [Google Scholar] [CrossRef]
- Parry, M.; Rose-Zerilli, M.J.; Gibson, J.; Ennis, S.; Walewska, R.; Forster, J.; Parker, H.; Davis, Z.; Gardiner, A.; Collins, A.; et al. Whole exome sequencing identifies novel recurrently mutated genes in patients with splenic marginal zone lymphoma. PLoS ONE 2013, 8, e83244. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Trifonov, V.; Fangazio, M.; Bruscaggin, A.; Rasi, S.; Spina, V.; Monti, S.; Vaisitti, T.; Arruga, F.; Fama, R.; et al. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH2 and other pathways regulating marginal zone development. J. Exp. Med. 2012, 209, 1537–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Song, B.; Guo, S.; Li, G.; Jin, G. Identification of germline and somatic mutations in pancreatic adenosquamous carcinoma using whole exome sequencing. Cancer Biomark 2020, 27, 389–397. [Google Scholar] [CrossRef]
- Wang, T.; Hoekzema, K.; Vecchio, D.; Wu, H.; Sulovari, A.; Coe, B.P.; Gillentine, M.A.; Wilfert, A.B.; Perez-Jurado, L.A.; Kvarnung, M.; et al. Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat. Commun. 2020, 11, 4932. [Google Scholar] [CrossRef]
- Zylicz, J.J.; Bousard, A.; Zumer, K.; Dossin, F.; Mohammad, E.; da Rocha, S.T.; Schwalb, B.; Syx, L.; Dingli, F.; Loew, D.; et al. The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell 2019, 176, 182–197.e123. [Google Scholar] [CrossRef] [Green Version]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Muller, C.W.; Vermeulen, M.; Muller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Cooper, S.; Grijzenhout, A.; Underwood, E.; Ancelin, K.; Zhang, T.; Nesterova, T.B.; Anil-Kirmizitas, B.; Bassett, A.; Kooistra, S.M.; Agger, K.; et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat. Commun. 2016, 7, 13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, J.; Zhang, Y.; Xu, R.M. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003, 17, 1823–1828. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Zhang, J.; Bonasio, R.; Strino, F.; Sawai, A.; Parisi, F.; Kluger, Y.; Reinberg, D. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 2012, 45, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Brown, J.L.; Cao, R.; Zhang, Y.; Kassis, J.A.; Jones, R.S. Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell 2004, 14, 637–646. [Google Scholar] [CrossRef]
- Chaligne, R.; Heard, E. X-chromosome inactivation in development and cancer. FEBS Lett. 2014, 588, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Pageau, G.J.; Hall, L.L.; Ganesan, S.; Livingston, D.M.; Lawrence, J.B. The disappearing Barr body in breast and ovarian cancers. Nat. Rev. Cancer 2007, 7, 628–633. [Google Scholar] [CrossRef]
- Brown, C.J.; Willard, H.F. The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature 1994, 368, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Csankovszki, G.; Panning, B.; Bates, B.; Pehrson, J.R.; Jaenisch, R. Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat. Genet. 1999, 22, 323–324. [Google Scholar] [CrossRef]
- Ross, M.T.; Grafham, D.V.; Coffey, A.J.; Scherer, S.; McLay, K.; Muzny, D.; Platzer, M.; Howell, G.R.; Burrows, C.; Bird, C.P.; et al. The DNA sequence of the human X chromosome. Nature 2005, 434, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Kain, M.; Wang, L. Inactivation of X-linked tumor suppressor genes in human cancer. Future Oncol. 2012, 8, 463–481. [Google Scholar] [CrossRef] [Green Version]
- Spatz, A.; Borg, C.; Feunteun, J. X-chromosome genetics and human cancer. Nat. Rev. Cancer 2004, 4, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef] [Green Version]
- Chaligne, R.; Popova, T.; Mendoza-Parra, M.A.; Saleem, M.A.; Gentien, D.; Ban, K.; Piolot, T.; Leroy, O.; Mariani, O.; Gronemeyer, H.; et al. The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res. 2015, 25, 488–503. [Google Scholar] [CrossRef] [Green Version]
- Jager, N.; Schlesner, M.; Jones, D.T.; Raffel, S.; Mallm, J.P.; Junge, K.M.; Weichenhan, D.; Bauer, T.; Ishaque, N.; Kool, M.; et al. Hypermutation of the inactive X chromosome is a frequent event in cancer. Cell 2013, 155, 567–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.; Dou, J.; Yang, G.; Chen, F. Long non-coding RNA XIST expression as a prognostic factor in human cancers: A meta-analysis. Int. J. Biol. Markers 2019, 34, 327–333. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Thomson, D.W.; Dinger, M.E. Endogenous microRNA sponges: Evidence and controversy. Nat. Rev. Genet. 2016, 17, 272–283. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.; Feng, L.; Li, F.; Sun, Z.; Wu, T.; Shi, X.; Li, J.; Li, X. Comprehensive characterization of lncRNA-mRNA related ceRNA network across 12 major cancers. Oncotarget 2016, 7, 64148–64167. [Google Scholar] [CrossRef] [Green Version]
- Madhi, H.; Kim, M.H. Beyond X-Chromosome Inactivation: The Oncogenic Facet of XIST in Human Cancers. Biomed. Sci. Lett. 2019, 25, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Giaimo, B.D.; Oswald, F.; Borggrefe, T. Dynamic chromatin regulation at Notch target genes. Transcription 2017, 8, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, N.T.; Su, H.; Khodadadi-Jamayran, A.; Lin, S.; Zhang, L.; Zhou, D.; Pawlik, K.M.; Townes, T.M.; Chen, Y.; Mulloy, J.C.; et al. The AS-RBM15 lncRNA enhances RBM15 protein translation during megakaryocyte differentiation. EMBO Rep. 2016, 17, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Zhang, J.; Breslin, P.; Onciu, M.; Ma, Z.; Morris, S.W. c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood 2009, 114, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Hiriart, E.; Gruffat, H.; Buisson, M.; Mikaelian, I.; Keppler, S.; Meresse, P.; Mercher, T.; Bernard, O.A.; Sergeant, A.; Manet, E. Interaction of the Epstein-Barr virus mRNA export factor EB2 with human Spen proteins SHARP, OTT1, and a novel member of the family, OTT3, links Spen proteins with splicing regulation and mRNA export. J. Biol. Chem. 2005, 280, 36935–36945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coker, H.; Wei, G.; Moindrot, B.; Mohammed, S.; Nesterova, T.; Brockdorff, N. The role of the Xist 5’ m6A region and RBM15 in X chromosome inactivation. Wellcome Open Res. 2020, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Newberry, E.P.; Latifi, T.; Towler, D.A. The RRM domain of MINT, a novel Msx2 binding protein, recognizes and regulates the rat osteocalcin promoter. Biochemistry 1999, 38, 10678–10690. [Google Scholar] [CrossRef]
- Oswald, F.; Kostezka, U.; Astrahantseff, K.; Bourteele, S.; Dillinger, K.; Zechner, U.; Ludwig, L.; Wilda, M.; Hameister, H.; Knochel, W.; et al. SHARP is a novel component of the Notch/RBP-Jkappa signalling pathway. EMBO J. 2002, 21, 5417–5426. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Vander-Wielen, B.D.; Giaimo, B.D.; Pan, L.; Collins, C.E.; Turkiewicz, A.; Hein, K.; Oswald, F.; Borggrefe, T.; Kovall, R.A. Structural and Functional Studies of the RBPJ-SHARP Complex Reveal a Conserved Corepressor Binding Site. Cell Rep. 2019, 26, 845–854.e846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, F.; Rodriguez, P.; Giaimo, B.D.; Antonello, Z.A.; Mira, L.; Mittler, G.; Thiel, V.N.; Collins, K.J.; Tabaja, N.; Cizelsky, W.; et al. A phospho-dependent mechanism involving NCoR and KMT2D controls a permissive chromatin state at Notch target genes. Nucleic Acids Res. 2016, 44, 4703–4720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oswald, F.; Winkler, M.; Cao, Y.; Astrahantseff, K.; Bourteele, S.; Knochel, W.; Borggrefe, T. RBP-Jkappa/SHARP recruits CtIP/CtBP corepressors to silence Notch target genes. Mol. Cell Biol. 2005, 25, 10379–10390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salat, D.; Liefke, R.; Wiedenmann, J.; Borggrefe, T.; Oswald, F. ETO, but not leukemogenic fusion protein AML1/ETO, augments RBP-Jkappa/SHARP-mediated repression of notch target genes. Mol. Cell Biol. 2008, 28, 3502–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, V.N.; Giaimo, B.D.; Schwarz, P.; Soller, K.; Vas, V.; Bartkuhn, M.; Blatte, T.J.; Dohner, K.; Bullinger, L.; Borggrefe, T.; et al. Heterodimerization of AML1/ETO with CBFbeta is required for leukemogenesis but not for myeloproliferation. Leukemia 2017, 31, 2491–2502. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, K.; Han, H.; Tani, S.; Tanigaki, K.; Tun, T.; Furukawa, T.; Taniguchi, Y.; Kurooka, H.; Hamada, Y.; Toyokuni, S.; et al. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity 2003, 18, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, M.; Shinkura, R.; Kuroda, K.; Yabe, D.; Honjo, T. Msx2-interacting nuclear target protein (Mint) deficiency reveals negative regulation of early thymocyte differentiation by Notch/RBP-J signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 1610–1615. [Google Scholar] [CrossRef] [Green Version]
- Sierra, O.L.; Cheng, S.L.; Loewy, A.P.; Charlton-Kachigian, N.; Towler, D.A. MINT, the Msx2 interacting nuclear matrix target, enhances Runx2-dependent activation of the osteocalcin fibroblast growth factor response element. J. Biol. Chem. 2004, 279, 32913–32923. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, J.; Qin, H.; Yang, H.; Li, J.; Zhou, P.; Liang, Y.; Han, H. Mint represses transactivation of the type II collagen gene enhancer through interaction with alpha A-crystallin-binding protein 1. J. Biol. Chem. 2005, 280, 18710–18716. [Google Scholar] [CrossRef] [Green Version]
- Giaimo, B.D.; Borggrefe, T. Introduction to Molecular Mechanisms in Notch Signal Transduction and Disease Pathogenesis. Adv. Exp. Med. Biol. 2018, 1066, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Giaimo, B.D.; Gagliani, E.K.; Kovall, R.A.; Borggrefe, T. Transcription Factor RBPJ as a Molecular Switch in Regulating the Notch Response. Adv. Exp. Med. Biol. 2021, 1287, 9–30. [Google Scholar] [CrossRef]
- McCarter, A.C.; Wang, Q.; Chiang, M. Notch in Leukemia. Adv. Exp. Med. Biol. 2018, 1066, 355–394. [Google Scholar] [CrossRef]
- Arieti, F.; Gabus, C.; Tambalo, M.; Huet, T.; Round, A.; Thore, S. The crystal structure of the Split End protein SHARP adds a new layer of complexity to proteins containing RNA recognition motifs. Nucleic Acids Res. 2014, 42, 6742–6752. [Google Scholar] [CrossRef] [Green Version]
- Schrodinger, LLC. The PyMOL. Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFbeta/BMP and hypoxia pathways. Biochim. Biophys. Acta 2016, 1863, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.Y.; Ordentlich, P.; Koyano-Nakagawa, N.; Tang, Z.; Downes, M.; Kintner, C.R.; Evans, R.M.; Kadesch, T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998, 12, 2269–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Hayward, S.D. Nuclear localization of CBF1 is regulated by interactions with the SMRT corepressor complex. Mol. Cell Biol. 2001, 21, 6222–6232. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Fujimuro, M.; Hsieh, J.J.; Chen, L.; Miyamoto, A.; Weinmaster, G.; Hayward, S.D. SKIP, a CBF1-associated protein, interacts with the ankyrin repeat domain of NotchIC To facilitate NotchIC function. Mol. Cell Biol. 2000, 20, 2400–2410. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Fujimuro, M.; Hsieh, J.J.; Chen, L.; Hayward, S.D. A role for SKIP in EBNA2 activation of CBF1-repressed promoters. J. Virol. 2000, 74, 1939–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepsen, K.; Solum, D.; Zhou, T.; McEvilly, R.J.; Kim, H.J.; Glass, C.K.; Hermanson, O.; Rosenfeld, M.G. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature 2007, 450, 415–419. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Choi, H.K.; Choi, K.C.; Park, S.Y.; Ota, I.; Yook, J.I.; Lee, Y.H.; Kim, K.; Yoon, H.G. Nuclear hormone receptor corepressor promotes esophageal cancer cell invasion by transcriptional repression of interferon-gamma-inducible protein 10 in a casein kinase 2-dependent manner. Mol. Biol. Cell 2012, 23, 2943–2954. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.Y.; Lim, B.J.; Choi, H.K.; Hong, S.W.; Jang, H.S.; Kim, C.; Chun, K.H.; Choi, K.C.; Yoon, H.G. CK2-NCoR signaling cascade promotes prostate tumorigenesis. Oncotarget 2013, 4, 972–983. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Gross, W.; Hong, S.H.; Privalsky, M.L. The SMRT corepressor is a target of phosphorylation by protein kinase CK2 (casein kinase II). Mol. Cell Biochem. 2001, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vander-Wielen, B.D.; Yuan, Z.; Friedmann, D.R.; Kovall, R.A. Transcriptional repression in the Notch pathway: Thermodynamic characterization of CSL-MINT (Msx2-interacting nuclear target protein) complexes. J. Biol. Chem. 2011, 286, 14892–14902. [Google Scholar] [CrossRef] [Green Version]
- Ariyoshi, M.; Schwabe, J.W. A conserved structural motif reveals the essential transcriptional repression function of Spen proteins and their role in developmental signaling. Genes Dev. 2003, 17, 1909–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knuckles, P.; Lence, T.; Haussmann, I.U.; Jacob, D.; Kreim, N.; Carl, S.H.; Masiello, I.; Hares, T.; Villasenor, R.; Hess, D.; et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018, 32, 415–429. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, J.; Yang, X.; Li, J.; Qin, H.; Dong, X.; Zhu, Y.; Liang, L.; Liang, Y.; Han, H. The Spen homolog Msx2-interacting nuclear target protein interacts with the E2 ubiquitin-conjugating enzyme UbcH8. Mol. Cell Biochem. 2006, 288, 151–157. [Google Scholar] [CrossRef]
- Li, J.; Li, J.; Yang, X.; Qin, H.; Zhou, P.; Liang, Y.; Han, H. The C terminus of MINT forms homodimers and abrogates MINT-mediated transcriptional repression. Biochim. Biophys. Acta 2005, 1729, 50–56. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lane, W.S.; Fischle, W.; Verdin, E.; Lazar, M.A.; Shiekhattar, R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000, 14, 1048–1057. [Google Scholar]
- Li, J.; Wang, J.; Wang, J.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, H.G.; Chan, D.W.; Huang, Z.Q.; Li, J.; Fondell, J.D.; Qin, J.; Wong, J. Purification and functional characterization of the human N-CoR complex: The roles of HDAC3, TBL1 and TBLR1. EMBO J. 2003, 22, 1336–1346. [Google Scholar] [CrossRef]
- Yoon, H.G.; Chan, D.W.; Reynolds, A.B.; Qin, J.; Wong, J. N-CoR mediates DNA methylation-dependent repression through a methyl CpG binding protein Kaiso. Mol. Cell 2003, 12, 723–734. [Google Scholar] [CrossRef]
- Wen, Y.D.; Perissi, V.; Staszewski, L.M.; Yang, W.M.; Krones, A.; Glass, C.K.; Rosenfeld, M.G.; Seto, E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2000, 97, 7202–7207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassi, M.T.; Ramesar, R.S.; Caciotti, B.; Winship, I.M.; de Grandi, A.; Riboni, M.; Townes, P.L.; Beighton, P.; Ballabio, A.; Borsani, G. X-linked late-onset sensorineural deafness caused by a deletion involving OA1 and a novel gene containing WD-40 repeats. Am. J. Hum. Genet. 1999, 64, 1604–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codina, A.; Love, J.D.; Li, Y.; Lazar, M.A.; Neuhaus, D.; Schwabe, J.W. Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2005, 102, 6009–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepsen, K.; Gleiberman, A.S.; Shi, C.; Simon, D.I.; Rosenfeld, M.G. Cooperative regulation in development by SMRT and FOXP1. Genes Dev. 2008, 22, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, K.; Hermanson, O.; Onami, T.M.; Gleiberman, A.S.; Lunyak, V.; McEvilly, R.J.; Kurokawa, R.; Kumar, V.; Liu, F.; Seto, E.; et al. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell 2000, 102, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrante, F.; Giaimo, B.D.; Bartkuhn, M.; Zimmermann, T.; Close, V.; Mertens, D.; Nist, A.; Stiewe, T.; Meier-Soelch, J.; Kracht, M.; et al. HDAC3 functions as a positive regulator in Notch signal transduction. Nucleic Acids Res. 2020, 48, 3496–3512. [Google Scholar] [CrossRef]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Muller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef] [Green Version]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef] [Green Version]
- You, S.H.; Lim, H.W.; Sun, Z.; Broache, M.; Won, K.J.; Lazar, M.A. Nuclear receptor co-repressors are required for the histone-deacetylase activity of HDAC3 in vivo. Nat. Struct. Mol. Biol. 2013, 20, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Barish, G.D.; Yu, R.T.; Karunasiri, M.S.; Becerra, D.; Kim, J.; Tseng, T.W.; Tai, L.J.; Leblanc, M.; Diehl, C.; Cerchietti, L.; et al. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab. 2012, 15, 554–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.F.; Melnick, A.; Lax, S.; Bouchard, D.; Liu, J.; Kiang, C.L.; Mayer, S.; Takahashi, S.; Licht, J.D.; Prive, G.G. Mechanism of SMRT corepressor recruitment by the BCL6 BTB domain. Mol. Cell 2003, 12, 1551–1564. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, W.; Gaudet, J.; Cheney, M.D.; Roudaia, L.; Cierpicki, T.; Klet, R.C.; Hartman, K.; Laue, T.M.; Speck, N.A.; et al. Structural basis for recognition of SMRT/N-CoR by the MYND domain and its contribution to AML1/ETO’s activity. Cancer Cell 2007, 11, 483–497. [Google Scholar] [CrossRef] [Green Version]
- Ghisletti, S.; Huang, W.; Jepsen, K.; Benner, C.; Hardiman, G.; Rosenfeld, M.G.; Glass, C.K. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes Dev. 2009, 23, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Lazar, M.A. The orphan nuclear receptor Rev-erbalpha recruits the N-CoR/histone deacetylase 3 corepressor to regulate the circadian Bmal1 gene. Mol. Endocrinol. 2005, 19, 1452–1459. [Google Scholar] [CrossRef]
- Malovannaya, A.; Lanz, R.B.; Jung, S.Y.; Bulynko, Y.; Le, N.T.; Chan, D.W.; Ding, C.; Shi, Y.; Yucer, N.; Krenciute, G.; et al. Analysis of the human endogenous coregulator complexome. Cell 2011, 145, 787–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikami, S.; Kanaba, T.; Ito, Y.; Mishima, M. NMR assignments of SPOC domain of the human transcriptional corepressor SHARP in complex with a C-terminal SMRT peptide. BioMol. NMR Assign. 2013, 7, 267–270. [Google Scholar] [CrossRef]
- Mikami, S.; Kanaba, T.; Takizawa, N.; Kobayashi, A.; Maesaki, R.; Fujiwara, T.; Ito, Y.; Mishima, M. Structural insights into the recruitment of SMRT by the corepressor SHARP under phosphorylative regulation. Structure 2014, 22, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Vadlamudi, R.K.; Manavathi, B.; Singh, R.R.; Nguyen, D.; Li, F.; Kumar, R. An essential role of Pak1 phosphorylation of SHARP in Notch signaling. Oncogene 2005, 24, 4591–4596. [Google Scholar] [CrossRef] [Green Version]
- Legare, S.; Cavallone, L.; Mamo, A.; Chabot, C.; Sirois, I.; Magliocco, A.; Klimowicz, A.; Tonin, P.N.; Buchanan, M.; Keilty, D.; et al. The Estrogen Receptor Cofactor SPEN Functions as a Tumor Suppressor and Candidate Biomarker of Drug Responsiveness in Hormone-Dependent Breast Cancers. Cancer Res. 2015, 75, 4351–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Bommer, G.T.; Zhai, Y.; Akyol, A.; Hinoi, T.; Winer, I.; Lin, H.V.; Cadigan, K.M.; Cho, K.R.; Fearon, E.R. Drosophila split ends homologue SHARP functions as a positive regulator of Wnt/beta-catenin/T-cell factor signaling in neoplastic transformation. Cancer Res. 2007, 67, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dansithong, W.; Jog, S.P.; Paul, S.; Mohammadzadeh, R.; Tring, S.; Kwok, Y.; Fry, R.C.; Marjoram, P.; Comai, L.; Reddy, S. RNA steady-state defects in myotonic dystrophy are linked to nuclear exclusion of SHARP. EMBO Rep. 2011, 12, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, N.; Koshimizu, E.; Okamoto, N.; Mizuno, S.; Ogata, T.; Nagai, T.; Kosho, T.; Ohashi, H.; Kato, M.; Sasaki, G.; et al. MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am. J. Med. Genet. A 2013, 161, 2234–2243. [Google Scholar] [CrossRef]
- Banka, S.; Veeramachaneni, R.; Reardon, W.; Howard, E.; Bunstone, S.; Ragge, N.; Parker, M.J.; Crow, Y.J.; Kerr, B.; Kingston, H.; et al. How genetically heterogeneous is Kabuki syndrome?: MLL2 testing in 116 patients, review and analyses of mutation and phenotypic spectrum. Eur. J. Hum. Genet. 2012, 20, 381–388. [Google Scholar] [CrossRef]
- Micale, L.; Augello, B.; Fusco, C.; Selicorni, A.; Loviglio, M.N.; Silengo, M.C.; Reymond, A.; Gumiero, B.; Zucchetti, F.; D’Addetta, E.V.; et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J. Rare Dis. 2011, 6, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulussen, A.D.; Stegmann, A.P.; Blok, M.J.; Tserpelis, D.; Posma-Velter, C.; Detisch, Y.; Smeets, E.E.; Wagemans, A.; Schrander, J.J.; van den Boogaard, M.J.; et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum. Mutat. 2011, 32, E2018–E2025. [Google Scholar] [CrossRef] [Green Version]
- Hannibal, M.C.; Buckingham, K.J.; Ng, S.B.; Ming, J.E.; Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.; Bigham, A.W.; Tabor, H.K.; Mefford, H.C.; et al. Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am. J. Med. Genet. A 2011, 155A, 1511–1516. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Bogershausen, N.; Alanay, Y.; Simsek-Kiper, P.O.; Plume, N.; Keupp, K.; Pohl, E.; Pawlik, B.; Rachwalski, M.; Milz, E.; et al. A mutation screen in patients with Kabuki syndrome. Hum. Genet. 2011, 130, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Makrythanasis, P.; van Bon, B.W.; Steehouwer, M.; Rodriguez-Santiago, B.; Simpson, M.; Dias, P.; Anderlid, B.M.; Arts, P.; Bhat, M.; Augello, B.; et al. MLL2 mutation detection in 86 patients with Kabuki syndrome: A genotype-phenotype study. Clin. Genet. 2013, 84, 539–545. [Google Scholar] [CrossRef]
- Miyake, N.; Mizuno, S.; Okamoto, N.; Ohashi, H.; Shiina, M.; Ogata, K.; Tsurusaki, Y.; Nakashima, M.; Saitsu, H.; Niikawa, N.; et al. KDM6A point mutations cause Kabuki syndrome. Hum. Mutat. 2013, 34, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.L.A.; Demarest, B.L.; Tone-Pah-Hote, T.; Tristani-Firouzi, M.; Yost, H.J. Inhibition of Notch signaling rescues cardiovascular development in Kabuki Syndrome. PLoS Biol. 2019, 17, e3000087. [Google Scholar] [CrossRef] [PubMed]
- Mercher, T.; Coniat, M.B.; Monni, R.; Mauchauffe, M.; Nguyen-Khac, F.; Gressin, L.; Mugneret, F.; Leblanc, T.; Dastugue, N.; Berger, R.; et al. Involvement of a human gene related to the Drosophila spen gene in the recurrent t(1;22) translocation of acute megakaryocytic leukemia. Proc. Natl. Acad. Sci. USA 2001, 98, 5776–5779. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Morris, S.W.; Valentine, V.; Li, M.; Herbrick, J.A.; Cui, X.; Bouman, D.; Li, Y.; Mehta, P.K.; Nizetic, D.; et al. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nat. Genet. 2001, 28, 220–221. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, S.; Zhang, Y.; Zhu, X. Biological effects of decreasing RBM15 on chronic myelogenous leukemia cells. Leuk Lymphoma 2012, 53, 2237–2244. [Google Scholar] [CrossRef]
- Kennison, J.A. Introduction to Trx-G and Pc-G genes. Methods EnzyMol. 2004, 377, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Wang, Y.; Jacobs, S.A.; Kim, Y.; Allis, C.D.; Khorasanizadeh, S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003, 17, 1870–1881. [Google Scholar] [CrossRef] [Green Version]
- Schertzer, M.D.; Braceros, K.C.A.; Starmer, J.; Cherney, R.E.; Lee, D.M.; Salazar, G.; Justice, M.; Bischoff, S.R.; Cowley, D.O.; Ariel, P.; et al. lncRNA-Induced Spread of Polycomb Controlled by Genome Architecture, RNA Abundance, and CpG Island DNA. Mol. Cell 2019, 75, 523–537.e510. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Gujar, H.; Weisenberger, D.J.; Liang, G. The Roles of Human DNA Methyltransferases and Their Isoforms in Shaping the Epigenome. Genes 2019, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.K.; Lawrie, C.H.; Green, T.M. Oncogenic Roles and Inhibitors of DNMT1, DNMT3A, and DNMT3B in Acute Myeloid Leukaemia. Biomark Insights 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Menafra, R.; Stunnenberg, H.G. MBD2 and MBD3: Elusive functions and mechanisms. Front. Genet. 2014, 5, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.P.; Skene, P.J.; Selfridge, J.; Clouaire, T.; Guy, J.; Webb, S.; Kerr, A.R.; Deaton, A.; Andrews, R.; James, K.D.; et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature 2010, 464, 1082–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voo, K.S.; Carlone, D.L.; Jacobsen, B.M.; Flodin, A.; Skalnik, D.G. Cloning of a mammalian transcriptional activator that binds unmethylated CpG motifs and shaRes. a CXXC domain with DNA methyltransferase, human trithorax, and methyl-CpG binding domain protein 1. Mol. Cell Biol. 2000, 20, 2108–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Possible causes of alteration of the Xi in cancer. Female somatic cells have one active and one inactive X chromosome, alterations of the Xi, often observed in cancer. These alterations can be caused by chromosome segregation errors, often leading to loss of the Xi and duplication of the Xa. Epigenetic erosion can lead to reactivation of X-linked genes. Mutations in the Xi happen more often than in other chromosomes. Abnormal Xist RNA levels are also observed in cancer. Xa = Active X chromosome, Xi = Inactive X chromosome.
Figure 1.
Possible causes of alteration of the Xi in cancer. Female somatic cells have one active and one inactive X chromosome, alterations of the Xi, often observed in cancer. These alterations can be caused by chromosome segregation errors, often leading to loss of the Xi and duplication of the Xa. Epigenetic erosion can lead to reactivation of X-linked genes. Mutations in the Xi happen more often than in other chromosomes. Abnormal Xist RNA levels are also observed in cancer. Xa = Active X chromosome, Xi = Inactive X chromosome.

Figure 2.
Proposed model for the silencing of the future inactive X chromosome (Xi) in female cells. The lncRNA Xist recruits SHARP [SMRT (silencing mediator for retinoid or thyroid hormone receptors) and HDACs (histone deacetylases)-associated repressor protein] to the X chromosome upon initiation of X chromosome inactivation (XCI). On one side, SHARP interacts with Xist through its RRMs (RNA recognition motifs) while on the other side it recruits chromatin modifiers through its highly conserved SPOC (Spen paralog and ortholog C-terminal) domain. One of the SPOC interactors is the multisubunit NCoR1/2 (nuclear receptor corepressor) complex that promotes histone deacetylation through its subunit HDAC3. As a next step, Xist interacts with hnRNPK (heterogeneous nuclear ribonucleoprotein K) recruiting the non-canonical PRC1 complex (ncPRC1) that writes H2AK119ub1 through RING1A/B (really interesting new gene 1 isoform A/B). Subsequently, H2AK119ub1 is recognized by JARID2 (jumanji and AT-rich interaction domain-containing 2), subunit of the PRC2 complex that writes H3K27me3 through EZH2/KMT6A [enhancer of zeste 2/lysine (K) methyltransferase 6A]. H3K27me3 is read by canonical PRC1 through its subunit CBX (chromobox-containing protein) and H2AK119ub1 is further established on the chromatin. Finally, silencing is achieved due to the activity of DNA methyltransferase 3B (DNMT3B) that methylates position 5 of cytosines (5 mC) within the DNA.
Figure 2.
Proposed model for the silencing of the future inactive X chromosome (Xi) in female cells. The lncRNA Xist recruits SHARP [SMRT (silencing mediator for retinoid or thyroid hormone receptors) and HDACs (histone deacetylases)-associated repressor protein] to the X chromosome upon initiation of X chromosome inactivation (XCI). On one side, SHARP interacts with Xist through its RRMs (RNA recognition motifs) while on the other side it recruits chromatin modifiers through its highly conserved SPOC (Spen paralog and ortholog C-terminal) domain. One of the SPOC interactors is the multisubunit NCoR1/2 (nuclear receptor corepressor) complex that promotes histone deacetylation through its subunit HDAC3. As a next step, Xist interacts with hnRNPK (heterogeneous nuclear ribonucleoprotein K) recruiting the non-canonical PRC1 complex (ncPRC1) that writes H2AK119ub1 through RING1A/B (really interesting new gene 1 isoform A/B). Subsequently, H2AK119ub1 is recognized by JARID2 (jumanji and AT-rich interaction domain-containing 2), subunit of the PRC2 complex that writes H3K27me3 through EZH2/KMT6A [enhancer of zeste 2/lysine (K) methyltransferase 6A]. H3K27me3 is read by canonical PRC1 through its subunit CBX (chromobox-containing protein) and H2AK119ub1 is further established on the chromatin. Finally, silencing is achieved due to the activity of DNA methyltransferase 3B (DNMT3B) that methylates position 5 of cytosines (5 mC) within the DNA.

Figure 3.
Structure modeling of SHARP RRM1. (A) Schematic representation of SHARP protein domains: RRM1 (aa 6 to 81), RRM1 (aa 335 to 410) RRM3 (aa 438 to 510), RRM4 (aa 517-587), Receptor Interaction Domain (RID, aa 2201 to 2707), RBP-J Interaction Domain (RBPID, aa 2776 to 2813), Spen Paralogue and Orthologue C-Terminal Domain (SPOC, aa 3417 to 3664). (B) Crystal structure of SHARP RRMs 2 to 4 (aa 335 to 620) from PDB: 4PQ6 [183]. (C) Homology model of SHARP (amino acids 6 to 81, accession NP_055816.2) performed with swissmodel (https://swissmodel.expasy.org/ (accessed date 14 January 2021). The model shows the typical β1-α1-β2-β3-α2-β4 topology of RRMs: Four antiparallel β sheets (yellow) framed from two α helices (red). Loops are shown in green. (D–F) Structural alignment of RRM1 (cyan) with RRM2 (D, yellow), RRM3 (E, orange), and RRM4 (F, red). Amino acids typically having contact with nucleic acids are highlighted. All of the structures and alignments were performed with PyMol [184].
Figure 3.
Structure modeling of SHARP RRM1. (A) Schematic representation of SHARP protein domains: RRM1 (aa 6 to 81), RRM1 (aa 335 to 410) RRM3 (aa 438 to 510), RRM4 (aa 517-587), Receptor Interaction Domain (RID, aa 2201 to 2707), RBP-J Interaction Domain (RBPID, aa 2776 to 2813), Spen Paralogue and Orthologue C-Terminal Domain (SPOC, aa 3417 to 3664). (B) Crystal structure of SHARP RRMs 2 to 4 (aa 335 to 620) from PDB: 4PQ6 [183]. (C) Homology model of SHARP (amino acids 6 to 81, accession NP_055816.2) performed with swissmodel (https://swissmodel.expasy.org/ (accessed date 14 January 2021). The model shows the typical β1-α1-β2-β3-α2-β4 topology of RRMs: Four antiparallel β sheets (yellow) framed from two α helices (red). Loops are shown in green. (D–F) Structural alignment of RRM1 (cyan) with RRM2 (D, yellow), RRM3 (E, orange), and RRM4 (F, red). Amino acids typically having contact with nucleic acids are highlighted. All of the structures and alignments were performed with PyMol [184].


{kind=link}
{kind=link}
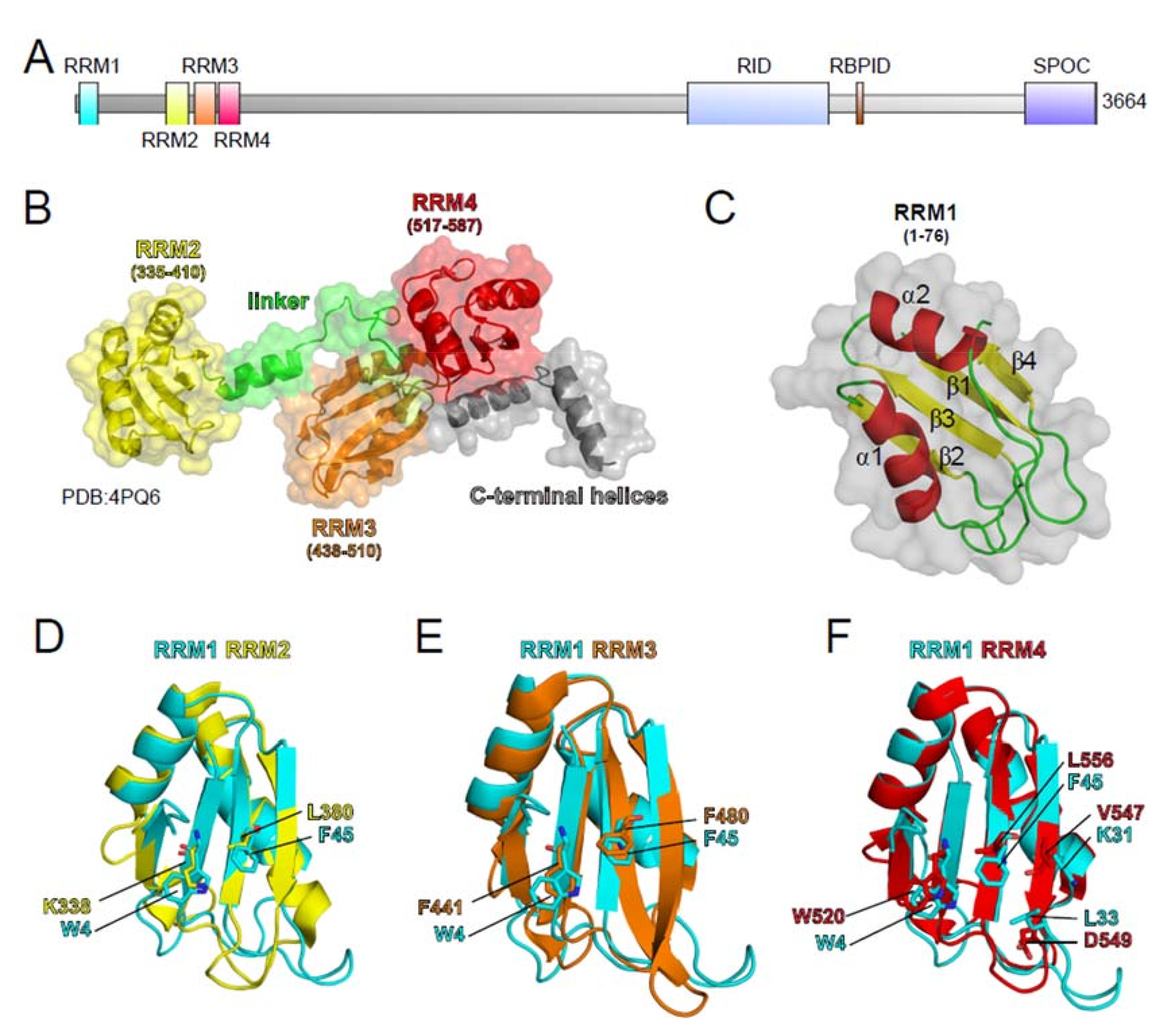{kind=link}
Table 1.
Proteins and complexes involved in the regulation of X chromosome inactivation (XCI). The “Disease(s)” column indicates diseases caused by mutations in the XCI related genes/proteins described in this table. The functional link between these mutations and XCI remains to be investigated.
Table 1.
Proteins and complexes involved in the regulation of X chromosome inactivation (XCI). The “Disease(s)” column indicates diseases caused by mutations in the XCI related genes/proteins described in this table. The functional link between these mutations and XCI remains to be investigated.
| Protein/Complex | Subunits | Function(s) in XCI | Disease(s) | References |
|---|---|---|---|---|
| DNMT3B | - | DNA methyltransferase | AML, FSHD, HD, ICF, PR | [56,57,58,59,60,61,62,63,64] |
| hnRNPK | - | Bridging protein between Xist and ncPRC1 | AKS, AML, KLS, KS, MF, OS | [65,66,67,68,69,70,71,72,73,74,75] |
| ncPRC1 | PCGF3/5 RING1A/B RYBP/YAF | E3 ubiquitin ligase | MDS | [65,76,77,78] |
| NCoR1/2 * | GPS2 HDAC3 NCoR1/2 * TBL1 TBLR1 | Deacetylase | ASDs, BC, CC, HCC, ID, MB, NDDs, OMZL, PS, SCZ | [79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95] |
| PRC1 | CBX2/4/6/7/8 PCGF1-6 PHC1-3 RING1A/B SCMH1/L2 | E3 ubiquitin ligase/Recognition of histone methylation | BC, DD, DSD, ESCC, GC, MCL, MDS, OSS, PM | [76,78,96,97,98,99,100,101,102,103,104,105] |
| PRC2 | AEBP2 EZH2 ** EED JARID2 RBBP4/7 SUZ12 | Methyltransferase | AML, DS-AMKL, DLBCL, ETP-ALL, FL, HCC, MDS, MPN, T-ALL, T-PLL | [76,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127] |
| SHARP | - | Adaptor protein that recruits the HDAC3-containing NCoR1/2 complexes | ACC, BC, DLBCL, MCL, NDDs, PASC, SMZL | [51,52,53,54,55,128,129,130,131,132,133,134,135,136,137,138,139] |
ACC: Adenoid cystic carcinoma; AEBP2: Adipocyte enhancer-binding protein 2; AKS: Au–Kline syndrome; AML: Acute myeloid leukemia; ASDs: Autism spectrum disorders; BC: Breast cancer; CC: Colon cancer; CBX2/4/6/7/8: Chromobox homolog 2/4/6/7/8; DD: Developmental disorder; DLBCL: Diffuse large B-cell lymphoma; DNMT3B: DNA methyltransferase 3B; DS-AMKL: Acute megakaryoblastic leukemia associated with Down syndrome; DSD: Disorders of sex development; EED: Embryonic ectoderm development; ESCC: Esophageal squamous cell carcinoma; ETP-ALL: Early T-cell precursor acute lymphoblastic leukaemia Early T-cell precursor acute lymphoblastic leukaemia; EZH2: Enhancer of zeste 2; FL: Follicular lymphoma; FSHD: Facioscapulohumeral dystrophy; GC: Gastric cancer; GPS2: G-protein pathway suppressor 2; HCC: Hepatocellular carcinoma; HD: Hirschsprung disease; HDAC3: Histone deacetylase 3; hnRNPK: Heterogeneous nuclear ribonucleoprotein K; ICF: Immunodeficiency, centromeric instability and facial anomalies; ID: Intellectual disability; JARID2: Jumanji and AT-rich interaction domain-containing 2; KLS: Kabuki-like syndrome; KS: Kabuki syndrome; MB: Medulloblastoma; MCL: Mantle cell lymphoma; MDS: Myelodysplastic syndromes; MF: Mycosis fungoides; MPN: myeloproliferative neoplasm; NCoR1/2: Nuclear receptor corepressor; ncPRC1: non-canonical PRC1 complex; NDDs: Neurodevelopmental disorders; OMZL: Ocular marginal zone lymphoma; OS: Okamoto syndrome; OSS: Osteosarcoma; PASC: Pancreatic adenosquamous carcinoma; PCGF1-6: PcG ring finger 1-6; PCGF3/5: PcG ring finger 3/5; PHC1-3: Polyhomeotic homolog 1-3; PM: Primary microcephaly; PR: Prostate cancer; PRC1: Polycomb repressive complex 1; PRC2: Polycomb repressive complex 2; PS: Pierpont syndrome; RBBP4/7: Retinoblastoma binding protein 4/7; RING1A/B: Really interesting new gene 1A/B; RYBP/YAF: RING1 And YY1 Binding Protein/YY1-associated factor; SCMH1/L2: Sex comb on midleg homolog 1/L2; SCZ: Schizophrenia; SHARP: SMRT (silencing mediator for retinoid or thyroid hormone receptors) and HDACs (histone deacetylases)-associated repressor protein; SMZL: Splenic marginal zone lymphoma; SUZ12: Suppressor of zeste 12; T-ALL: T-cell acute lymphoblastic leukemia; T-PLL: T-cell prolymphocytic leukemia; TBL1: Transducin β-like protein 1; TBLR1: Transducing β-like 1 (TBL1)-related protein; XCI: X chromosome inactivation; Xist: X inactive specific transcript; * NCoR2 is also known as SMRT (silencing mediator for retinoid or thyroid hormone receptors); ** EZH2 is also known as KMT6A (lysine (K) methyltransferase 6A).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Giaimo, B.D.; Robert-Finestra, T.; Oswald, F.; Gribnau, J.; Borggrefe, T. Chromatin Regulator SPEN/SHARP in X Inactivation and Disease. Cancers 2021, 13, 1665. https://doi.org/10.3390/cancers13071665
AMA Style
Giaimo BD, Robert-Finestra T, Oswald F, Gribnau J, Borggrefe T. Chromatin Regulator SPEN/SHARP in X Inactivation and Disease. Cancers. 2021; 13(7):1665. https://doi.org/10.3390/cancers13071665
Chicago/Turabian StyleGiaimo, Benedetto Daniele, Teresa Robert-Finestra, Franz Oswald, Joost Gribnau, and Tilman Borggrefe. 2021. "Chromatin Regulator SPEN/SHARP in X Inactivation and Disease" Cancers 13, no. 7: 1665. https://doi.org/10.3390/cancers13071665
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.