
Antibody-Drug Conjugates in Urothelial Carcinoma: A New Therapeutic Opportunity Moves from Bench to Bedside

 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Antibody-Drug Conjugates
2.1. Identification of a Suitable Antibody
2.2. Linker
2.3. Payloads
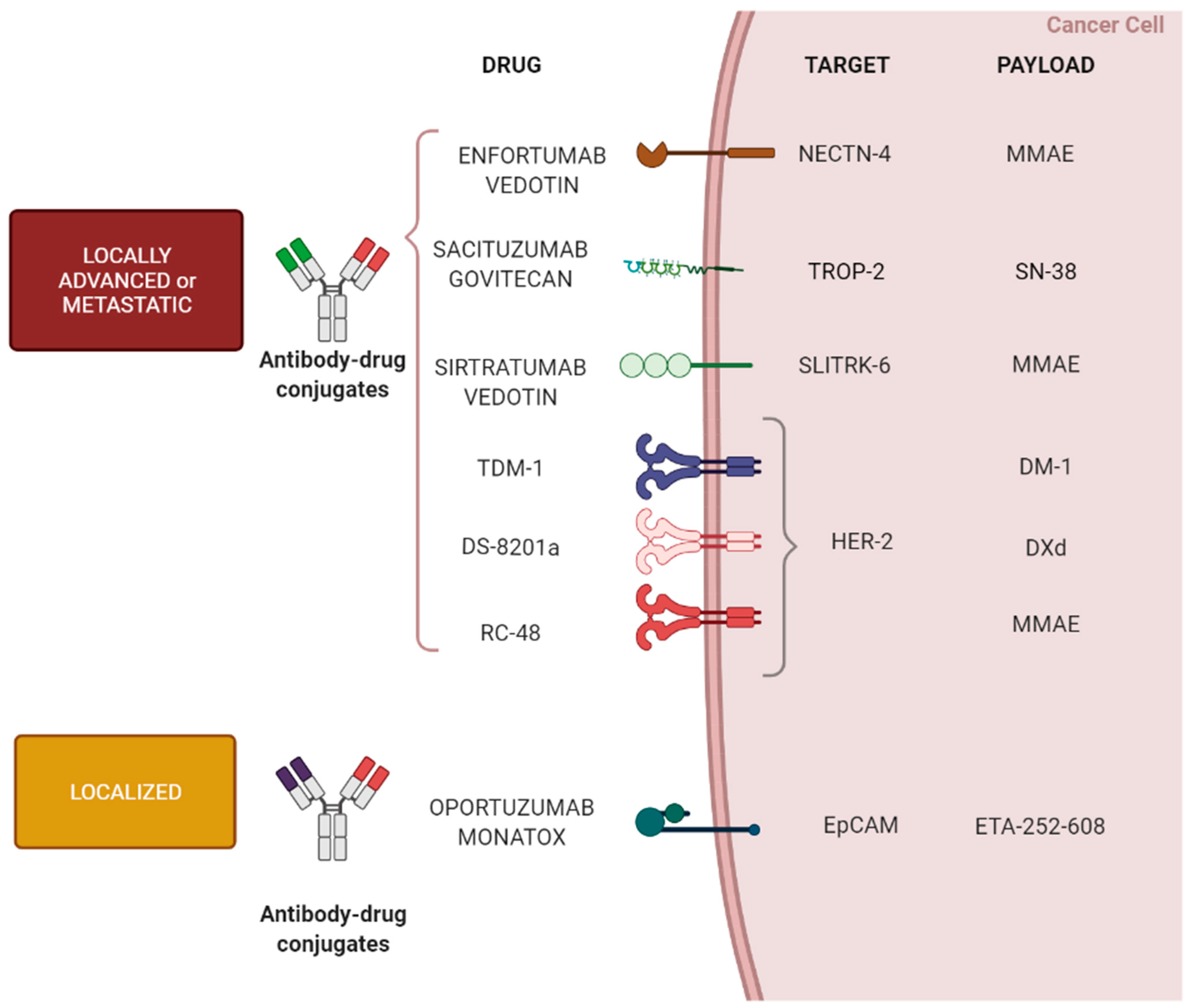
3. ADC Anti-Tumor Activity in Urothelial Carcinoma
3.1. Metastatic Setting
3.1.1. Enfortumab Vedotin
3.1.2. Sacituzumab Govitecan
3.1.3. Sirtratumab Vedotin (ASG15-ME)
3.1.4. Targeting HER-2 in Bladder Cancer
3.1.4.1. Trastuzumab Emtansine (TDM-1)
3.1.4.2. Trastuzumab Deruxtecan (DS-8201a)
3.1.4.3. Disitamab Vedotin (RC-48)
3.1.4.4. Drug-Conjugates beyond Antibodies
3.2. Activity of ADC in Localized Bladder Cancer
Oportuzumab Monatox
4. Potential Mechanism of Resistance
4.1. Lack of Antigen Attachment
4.2. Suppression of Payload Efficacy
4.3. Cell Cycle Alterations
4.4. Alteration in Trafficking Pathways
5. Role of ADCs in the Therapeutic Sequence of Advanced UC
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A.; Compérat, E.; Cowan, N.C.; De Santis, M.; Gakis, G.; James, N.; Lebrét, T.; Sherif, A.; van der Heijden, A.G.; Ribal, M.J. Muscle-invasive and metastatic bladder cancer. Eur. Urol. Guidel. 2015, 71, 462–475. [Google Scholar]
- Fuge, O.; Vasdev, N.; Allchorne, P.; Green, J.S. Immunotherapy for bladder cancer. Res. Rep. Urol. 2015, 7, 65. [Google Scholar]
- Babjuk, M.; Oosterlinck, W.; Sylvester, R.; Kaasinen, E.; Böhle, A.; Palou-Redorta, J.; Rouprêt, M. EAU guidelines on non–muscle-invasive urothelial carcinoma of the bladder, the 2011 update. Eur. Urol. 2011, 59, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Kamat, A.M.; Kulkarni, G.S.; Uchio, E.M.; Boormans, J.L.; Roumiguié, M.; Krieger, L.E.M.; Singer, E.A.; Bajorin, D.F.; Grivas, P.; et al. Pembrolizumab monotherapy for the treatment of high-risk non-muscle-invasive bladder cancer unre-sponsive to BCG (KEYNOTE-057): An open-label, single-arm, multicentre, phase 2 study. Lancet Oncol. 2021. [CrossRef]
- Funt, S.A.; Rosenberg, J.E. Systemic, perioperative management of muscle-invasive bladder cancer and future horizons. Nat. Rev. Clin. Oncol. 2017, 14, 221–234. [Google Scholar] [CrossRef]
- von der Maase, H.; Hansen, S.W.; Roberts, J.T.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Bodrogi, I.; Albers, P.; Knuth, A.; Lippert, C.M.; et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: Results of a large, randomized, multinational, multicenter, phase III study. J. Clin. Oncol. 2000, 18, 3068–3077. [Google Scholar] [CrossRef]
- Flaig, T.W.; Spiess, P.E.; Agarwal, N.; Bangs, R.; Boorjian, S.A.; Buyyounouski, M.K.; Chang, S.; Downs, T.M.; Efstathiou, J.A.; Friedlander, T.; et al. Bladder Cancer, Version 3.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2020, 18, 329–354. [Google Scholar] [CrossRef] [Green Version]
- Galsky, M.D.; Hahn, N.M.; Rosenberg, J.E.; Sonpavde, G.; Hutson, T.; Oh, W.K.; Dreicer, R.; Vogelzang, N.J.; Sternberg, C.N.; Bajorin, D.F.; et al. Treatment of Patients With Metastatic Urothelial Cancer “Unfit” for Cisplatin-Based Chemotherapy. J. Clin. Oncol. 2011, 29, 2432–2438. [Google Scholar] [CrossRef] [Green Version]
- NCCN Guidelines for Bladder Cancer, V.5.2021. Available online: https://www.nccn.org/guidelines/guidelines-process/transparency-process-and-recommendations/GetFileFromFileManager?fileManagerId=12952 (accessed on 24 September 2021).
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef]
- Parikh, M.; Powles, T. Immune Checkpoint Inhibition in Advanced Bladder and Kidney Cancer: Responses and Further Management. Am. Soc. Clin. Oncol. Educ. Book 2021, 41, 182–189. [Google Scholar] [CrossRef]
- Oing, C.; Rink, M.; Oechsle, K.; Seidel, C.; von Amsberg, G.; Bokemeyer, C. Second Line Chemotherapy for Advanced and Metastatic Urothelial Carcinoma: Vinflunine and Beyond—A Comprehensive Review of the Current Literature. J. Urol. 2015, 195, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, B.; Siefker-Radtke, A.O.; Srinivas, S.; Yu, E.Y. Systemic Therapy for Advanced Urothelial Carcinoma: Current Standards and Treatment Considerations. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, A.; de Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody–drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef]
- Sliwkowski, M.X.; Mellman, I. Antibody Therapeutics in Cancer. Science 2013, 341, 1192–1198. [Google Scholar] [CrossRef]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody–drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). OncoImmunology 2017, 7, e1395127. [Google Scholar] [CrossRef]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed]
- Schuurman, J.; Parren, P.W. Editorial overview: Special section: New concepts in antibody therapeutics: What’s in store for antibody therapy? Curr. Opin. Immunol. 2016, 40, 52–103. [Google Scholar] [CrossRef] [PubMed]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiller, K.E.; Tessier, P.M. Advances in Antibody Design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Song, Y.; Tian, W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 2020, 13, 45. [Google Scholar] [CrossRef]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody–drug conjugates: The nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [Green Version]
- Polson, A.G.; Ho, W.Y.; Ramakrishnan, V. Investigational antibody-drug conjugates for hematological malignancies. Expert Opin. Investig. Drugs 2010, 20, 75–85. [Google Scholar] [CrossRef]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Lucas, A.T.; Price, L.S.L.; Schorzman, A.N.; Storrie, M.; Piscitelli, J.A.; Razo, J.; Zamboni, W.C. Factors Affecting the Pharmacology of Antibody–Drug Conjugates. Antibodies 2018, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of drug–antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody–maytansinoid conjugates. Bioconjugate Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef]
- King, H.D.; Dubowchik, G.M.; Mastalerz, H.; Willner, D.; Hofstead, S.J.; Firestone, R.A.; Lasch, S.J.; Trail, P.A. Monoclonal Antibody Conjugates of Doxorubicin Prepared with Branched Peptide Linkers: Inhibition of Aggregation by Methoxytriethyleneglycol Chains. J. Med. Chem. 2002, 45, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R. The Immunogenicity of Humanized and Fully Human Antibodies: Residual Immunogenicity Resides in the CDR Regions. Available online: https://pubmed.ncbi.nlm.nih.gov/20400861 (accessed on 1 May 2010).
- Hwang, W.Y.K.; Foote, J. Immunogenicity of engineered antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S. Immunogenicity of biological therapeutics: A hierarchy of concerns. Dev. Biol. 2003, 112, 15–21. [Google Scholar]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [Green Version]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2016, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Drake, P.M.; Rabuka, D. An emerging playbook for antibody–drug conjugates: Lessons from the laboratory and clinic suggest a strategy for improving efficacy and safety. Curr. Opin. Chem. Biol. 2015, 28, 174–180. [Google Scholar] [CrossRef]
- Ducry, L.; Stump, B. Antibody-Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjugate Chem. 2009, 21, 5–13. [Google Scholar] [CrossRef]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjugate Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Paul, S.; Kantarjian, H. The clinical development of antibody–drug conjugates—Lessons from leukaemia. Nat. Rev. Clin. Oncol. 2021, 18, 418–433. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers Having a Crucial Role in Antibody–Drug Conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V. Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.N.; Sugarman, S.; Murray, J.; Ostroff, J.B.; Healey, D.; Jones, D.; Daniel, C.R.; Lebherz, D.; Brewer, H.; Onetto, N.; et al. Phase I Trial of the Anti–Lewis Y Drug Immunoconjugate BR96-Doxorubicin in Patients With Lewis Y–Expressing Epithelial Tumors. J. Clin. Oncol. 2000, 18, 2282–2292. [Google Scholar] [CrossRef]
- Kanellos, J.; Pietersz, G.A.; McKenzie, I.F.C. Studies of Methotrexate-Monoclonal Antibody Conjugates for Immunotherapy. JNCI: J. Natl. Cancer Inst. 1985, 75, 319–332. [Google Scholar] [CrossRef]
- Starling, J.J.; Maciak, R.S.; Law, K.L.; A Hinson, N.; Briggs, S.L.; Laguzza, B.C.; A Johnson, D. In vivo antitumor activity of a monoclonal antibody-Vinca alkaloid immunoconjugate directed against a solid tumor membrane antigen characterized by heterogeneous expression and noninternalization of antibody-antigen complexes. Cancer Res. 1991, 51, 2965–2972. [Google Scholar]
- Trail, P.A.; Willner, D.; Lasch, S.J.; Henderson, A.J.; Hofstead, S.; Casazza, A.M.; Firestone, R.A.; Hellström, I.; Hellström, K.E. Cure of Xenografted Human Carcinomas by BR96-Doxorubicin Immunoconjugates. Science 1993, 261, 212–215. [Google Scholar] [CrossRef]
- Hoffmann, D.; Lorenz, P.; Bosslet, K.; Seemann, G.; Czech, J.; Kolar, C.; Sedlacek, H. Antibodies as Carriers of Cytotoxicity. Contrib. Oncol. 1992, 43, 1–10. [Google Scholar] [CrossRef]
- Mach, J.P.; Carrel, S.; Forni, M.; Ritschard, J.; Donath, A.; Alberto, P. Tumor localization of radio-labeled antibodies against carcinoembryonic antigen in patients with carcinoma: A critical evaluation. N. Engl. J. Med. 1980, 303, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.H.; Jung, I.H. Modeling to capture bystander-killing effect by released payload in target positive tumor cells. BMC Cancer 2019, 19, 194. [Google Scholar] [CrossRef] [PubMed]
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.; Sussman, D.; Prota, A.E. Structural Basis of Microtubule Destabilization by Potent Auristatin Anti-Mitotics. PLoS ONE 2016, 11, e0160890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yardley, D.A.; Krop, I.E.; LoRusso, P.M.; Mayer, M.; Barnett, B.; Yoo, B.; Perez, E.A. Trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer previously treated with chemotherapy and 2 or more HER2-targeted agents: Results from the T-PAS expanded access study. Cancer J. 2015, 21, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Zein, N.; Sinha, A.M.; McGahren, W.J.; Ellestad, G.A. Calicheamicin γ1I: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 1988, 240, 1198–1201. [Google Scholar] [CrossRef]
- Smellie, M.; Kelland, L.; Thurston, D.; Souhami, R.; Hartley, J. Cellular pharmacology of novel C8-linked anthramycin-based sequence-selective DNA minor groove cross-linking agents. Br. J. Cancer 1994, 70, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Hurley, L.H.; Reck, T.; Thurston, D.E.; Langley, D.R.; Holden, K.G.; Hertzberg, R.P.; Hoover, J.R.E.; Gallagher, G.; Faucette, L.F. Pyrrolo-1,4-benzodiazepine antitumor antibiotics: Relationship of DNA alkylation and sequence specificity to the biological activity of natural and synthetic compounds. Chem. Res. Toxicol. 1988, 1, 258–268. [Google Scholar] [CrossRef]
- Jenkins, T.C.; Hurley, L.H.; Neidle, S.; Thurston, D.E. Structure of a Covalent DNA Minor Groove Adduct with a Pyrrolobenzodiazepine Dimer: Evidence for Sequence-Specific Interstrand Crosslinking. J. Med. Chem. 1994, 37, 4529–4537. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Cardillo, T.M.; Govindan, S.V.; Rossi, E.A.; Sharkey, R.M. Correction: Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget 2020, 11, 942. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnell, R.; Staak, O.; Borchmann, P.; Schwartz, C.; Matthey, B.; Hansen, H.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Diehl, V.; et al. Phase I study with an anti-CD30 ricin A-chain immunotoxin (Ki-4. dgA) in patients with refractory CD30+ Hodgkin’s and non-Hodgkin’s lymphoma. Clin. Cancer Res. 2002, 8, 1779–1786. [Google Scholar] [PubMed]
- Madhumathi, J.; Devilakshmi, S.; Sridevi, S.; Verma, R.S. Immunotoxin therapy for hematologic malignancies: Where are we heading? Drug Discov. 2016, 21, 325–332. [Google Scholar] [CrossRef]
- Akbari, B.; Farajnia, S.; Ahdi Khosroshahi, S.; Safari, F.; Yousefi, M.; Dariushnejad, H.; Rahbarnia, L. Immunotoxins in cancer therapy: Review and update. Int. Rev. Immunol. 2017, 36, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Reymond, N.; Fabre, S.; Lecocq, E.; Adelaıde, J.; Dubreuil, P.; Lopez, M. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J. Biol. Chem. 2001, 276, 43205–43215. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, S.T.; Faltas, B.M.; Lam, E.T.; Saylor, P.J.; Bardia, A.; Hajdenberg, J.; Morgans, A.K.; Lim, E.A.; Kalinsky, K.; Simpson, P.S.; et al. Sacituzumab govitecan (IMMU-132) in patients with previously treated metastatic urothelial cancer (mUC): Results from a phase I/II study. J. Clin. Oncol. 2019, 37, 354. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- Heath, E.I.; Rosenberg, J.E. The biology and rationale of targeting nectin-4 in urothelial carcinoma. Nat. Rev. Urol. 2020, 18, 93–103. [Google Scholar] [CrossRef]
- Kedashiro, S.; Sugiura, A.; Mizutani, K.; Takai, Y. Nectin-4 cis-interacts with ErbB2 and its trastuzumab-resistant splice variants, enhancing their activation and DNA synthesis. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.; Sridhar, S.S.; Zhang, J.; Smith, D.; Ruether, D.; Flaig, T.W.; Baranda, J.; Lang, J.; Plimack, E.R.; Sangha, R.; et al. EV-101: A phase I study of single-agent enfortumab vedotin in patients with nectin-4–positive solid tumors, including metastatic urothelial carcinoma. J. Clin. Oncol. 2020, 38, 1041. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2019, 37, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. FDA Approves New Type of Therapy to Treat Advanced Urothelial Cancer. Available online: https//www.fda.gov/news-events/press-Announcurothelial-cancer (accessed on 30 December 2019).
- Hoimes, C.J.; Rosenberg, J.E.; Petrylak, D.P.; Carret, A.-S.; Sasse, C.; Chaney, M.F.; Flaig, T.W. Study EV-103: New cohorts testing enfortumab vedotin alone or in combination with pembrolizumab in muscle invasive urothelial cancer. J. Clin. Oncol. 2020, 38, 595. [Google Scholar] [CrossRef]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Padcev (enfortumab vedotin-ejfv) Prescribing Information, Astellas Pharma. Available online: www.accessdata.fda.gov (accessed on 30 January 2020).
- Cardillo, T.M.; Govindan, S.V.; Sharkey, R.M.; Trisal, P.; Goldenberg, D.M. Humanized anti-Trop-2 IgG-SN-38 conjugate for effective treatment of diverse epithelial cancers: Preclinical studies in human cancer xenograft models and monkeys. Clin. Cancer Res. 2011, 17, 3157–3169. [Google Scholar] [CrossRef] [Green Version]
- Cubas, R.; Zhang, S.; Li, M.; Chen, C.; Yao, Q. Trop2 expression contributes to tumor pathogenesis by activating the ERK MAPK pathway. Mol. Cancer 2010, 9, 253. [Google Scholar] [CrossRef] [Green Version]
- Avellini, C.; Licini, C.; Lazzarini, R.; Gesuita, R.; Guerra, E.; Tossetta, G.; Castellucci, C.; Giannubilo, S.R.; Procopio, A.; Alberti, S.; et al. The trophoblast cell surface antigen 2 and miR-125b axis in urothelial bladder cancer. Oncotarget 2017, 8, 58642–58653. [Google Scholar] [CrossRef] [Green Version]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Guarino, M.J.; Picozzi, J.V.J.; Vahdat, L.T.; Thomas, S.S.; Govindan, S.V.; Maliakal, P.P.; Wegener, W.A.; et al. First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clin. Cancer Res. 2015, 21, 3870–3878. [Google Scholar] [CrossRef] [Green Version]
- Tagawa, S.T.; Balar, A.V.; Petrylak, D.P.; Kalebasty, A.R.; Loriot, Y.; Fléchon, A.; Jain, R.K.; Agarwal, N.; Bupathi, M.; Barthelemy, P.; et al. TROPHY-U-01: A Phase II Open-Label Study of Sacituzumab Govitecan in Patients With Metastatic Urothelial Carcinoma Progressing After Platinum-Based Chemotherapy and Checkpoint Inhibitors. J. Clin. Oncol. 2021, 39, 2474–2485. [Google Scholar] [CrossRef]
- Morrison, K.; Challita-Eid, P.M.; Raitano, A.; An, Z.; Yang, P.; Abad, J.D.; Liu, W.; Lortie, D.R.; Snyder, J.T.; Capo, L.; et al. Development of ASG-15ME, a novel antibody–drug conjugate targeting SLITRK6, a new urothelial cancer biomarker. Mol. Cancer Ther. 2016, 15, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morlet, T.; Rabinowitz, M.R.; Looney, L.R.; Riegner, T.; Greenwood, L.A.; Sherman, E.A.; Achilly, N.; Zhu, A.; Yoo, E.; O’Reilly, R.C.; et al. A homozygous SLITRK6 nonsense mutation is associated with progressive auditory neuropathy in humans. Laryngoscope 2013, 124, E95–E103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrylak, D.; Heath, E.; Sonpavde, G.; George, S.; Morgans, A.; Eigl, B.; Picus, J.; Cheng, S.; Hotte, S.; Gartner, E.; et al. Interim analysis of a phase I dose escalation trial of the antibody drug conjugate (ADC) AGS15E (ASG-15ME) in patients (Pts) with metastatic urothelial cancer (mUC). Ann. Oncol. 2016, 27, vi269. [Google Scholar] [CrossRef] [Green Version]
- Klapper, L.N.; Waterman, H.; Sela, M.; Yarden, Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res. 2000, 60, 3384–3388. [Google Scholar] [PubMed]
- Oh, D.-Y.; Bang, Y.-J. HER2-targeted therapies —A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2019, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Schwaederle, M.; Arguello, D.; Millis, S.Z.; Gatalica, Z.; Kurzrock, R. HER2 expression status in diverse cancers: Review of results from 37,992 patients. Cancer Meta. Rev. 2015, 34, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Kiss, B.; Wyatt, A.W.; Douglas, J.; Skuginna, V.; Mo, F.; Anderson, S.; Rotzer, D.; Fleischmann, A.; Genitsch, V.; Hayashi, T.; et al. Her2 alterations in muscle-invasive bladder cancer: Patient selection beyond protein expression for targeted therapy. Sci. Rep. 2017, 7, 42713. [Google Scholar] [CrossRef]
- Krüger, S.; Weitsch, G.; Büttner, H.; Matthiensen, A.; Böhmer, T.; Marquardt, T.; Sayk, F.; Feller, A.C.; Böhle, A. HER2 overexpression in muscle-invasive urothelial carcinoma of the bladder: Prognostic implications. Int. J. Cancer 2002, 102, 514–518. [Google Scholar] [CrossRef]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J.; et al. HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Seiler, R.; Oo, H.Z.; Jäger, W.; Moskalev, I.; Awrey, S.; Dejima, T.; Todenhöfer, T.; Li, N.; Fazli, L.; et al. Targeting HER2 with T-DM1, an Antibody Cytotoxic Drug Conjugate, is Effective in HER2 Over Expressing Bladder Cancer. J. Urol. 2015, 194, 1120–1131. [Google Scholar] [CrossRef]
- Li, B.T.; Makker, V.; Buonocore, D.J.; Offin, M.D.; Olah, Z.T.; Panora, E.; Shen, R.; Ho, A.L.; Yaeger, R.; Iyer, G.; et al. A multi-histology basket trial of ado-trastuzumab emtansine in patients with HER2 amplified cancers. J. Clin. Oncol. 2018, 36, 2502. [Google Scholar] [CrossRef]
- Modi, S.; Tsurutani, J.; Tamura, K.; Park, H.; Sagara, Y.; Murthy, R.; Iwata, H.; Krop, I.; Doi, T.; Redfern, C.; et al. Abstract P6-17-02: Trastuzumab deruxtecan (DS-8201a) in subjects with HER2-low expressing breast cancer: Updated results of a large phase 1 study. Cancer Res. 2019, 79, P6-17-02. [Google Scholar] [CrossRef]
- Daiichi Sankyo Inc. Trastuzumab Deruxtecan (DS-8201a) With Nivolumab in Advanced Breast and Urothelial Cancer NCT03523572. 2022. Available online: https//clinicaltrials.gov/ct2/show/ (accessed on 15 January 2022).
- Rinnerthaler, G.; Gampenrieder, S.P.; Greil, R. HER2 Directed Antibody-Drug-Conjugates beyond T-DM1 in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 1115. [Google Scholar] [CrossRef] [Green Version]
- Sheng, X.; Yan, X.; Wang, L.; Shi, Y.-X.; Yao, X.; Luo, H.; Shi, B.; Liu, J.-Y.; He, Z.; Yu, G.; et al. Open-label, Multicenter, Phase II Study of RC48-ADC, a HER2-Targeting Antibody–Drug Conjugate, in Patients with Locally Advanced or Metastatic Urothelial Carcinoma. Clin. Cancer Res. 2020, 27, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Nilvebrant, J.; Nygren, P.; Lehmann, F. Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy. Molecules 2021, 26, 6042. [Google Scholar] [CrossRef] [PubMed]
- Karasseva, N.G.; Glinsky, V.; Chen, N.X.; Komatireddy, R.; Quinn, T.P. Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J. Protein Chem. 2002, 21, 287–296. [Google Scholar] [CrossRef]
- Adorján, A.E.; Bósze, S.; Szabó, I.; Mezó, G. Structure-activity relationship of HER2 receptor targeting peptide and its derivatives in targeted tumor therapy. Biomolecules 2020, 10, 183. [Google Scholar] [CrossRef]
- Pramanik, A.; Laha, D.; Dash, S.K.; Chattopadhyay, S.; Roy, S.; Das, D.K.; Pramanik, P.; Karmakar, P. An in-vivo study for targeted delivery of copper-organic complex to breast cancer using chitosan polymer nanoparticles. Mater. Sci. Eng. C 2016, 68, 327–337. [Google Scholar] [CrossRef]
- Evan, Y.Y.; Petrylak, D.P.; O’Donnell, P.H.; Lee, J.L.; van der Heijden, M.S.; Loriot, Y.; Stein, M.N.; Necchi, A.; Kojima, T.; Harrison, M.R.; et al. Enfortumab vedotin after PD-1 or PD-L1 inhibitors in cisplatin-ineligible patients with advanced urothelial carcinoma (EV-201): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 872–882. [Google Scholar]
- Bardia, A.; Messersmith, W.; Kio, E.; Berlin, J.; Vahdat, L.; Masters, G.; Moroose, R.; Santin, A.; Kalinsky, K.; Picozzi, V.; et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: Final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann. Oncol. 2021, 32, 746–756. [Google Scholar] [CrossRef]
- Sheng, X.; Zhou, A.-P.; Yao, X.; Shi, Y.; Luo, H.; Shi, B.; Liu, J.; Yu, G.; He, Z.; Hu, C.; et al. A phase II study of RC48-ADC in HER2-positive patients with locally advanced or metastatic urothelial carcinoma. J. Clin. Oncol. 2019, 37, 4509. [Google Scholar] [CrossRef]
- Eyvazi, S.; Farajnia, S.; Dastmalchi, S.; Kanipour, F.; Zarredar, H.; Bandehpour, M.; Bandehpour, M. Antibody Based EpCAM Targeted Therapy of Cancer, Review and Update. Curr. Cancer Drug Targets 2018, 18, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Spizzo, G.; Fong, D.; Wurm, M.; Ensinger, C.; Obrist, P.; Hofer, C.; Mazzoleni, G.; Gastl, G.; Went, P. EpCAM expression in primary tumour tissues and metastases: An immunohistochemical analysis. J. Clin. Pathol. 2011, 64, 415–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Martin, R.C.G.; Zheng, Q.; Farmer, R.; Pandit, H.; Li, X.; Jacob, K.; Suo, J.; Li, Y. Drug-induced expression of EpCAM contributes to therapy resistance in esophageal adenocarcinoma. Cell. Oncol. 2018, 41, 651–662. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, G.C.; Rasamoelisolo, M.; Entwistle, J.; Cizeau, J.; Bosc, D.; Cuthbert, W.; Kowalski, M.; Spearman, M.; Glover, N. A phase I clinical study of VB4-845: Weekly intratumoral administration of an anti-EpCAM recombinant fusion protein in patients with squamous cell carcinoma of the head and neck. Drug Des. Devel. Ther. 2008, 2, 105. [Google Scholar]
- Liu, Y.; Wu, R.; Gavrilescu, C.; Sagert, J.; Tipton, K.; Liu, S.; Chan, C.; Boulé, S.; Wilhelm, A.; Lucas, J.; et al. Abstract 213: Development of a Probody-Drug Conjugate Targeting EpCAM for the Treatment of Solid Tumors. 2019. Available online: https://doi.org/10.1158/1538-7445.am2019-213 (accessed on 24 September 2021).
- MacDonald, G.C.; Kowalski, M.; Entwistle, J.; Cizeau, J.; Niforos, D.; Loewen, S.; Chapman, W. A Phase I study of an intravesically administered immunotoxin targeting EpCAM for the treatment of nonmuscle-invasive bladder cancer in BCG-refractory and BCG-intolerant patients. Drug Des. Dev. Ther. 2010, 4, 313–320. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, M.; Guindon, J.; Brazas, L.; Moore, C.; Entwistle, J.; Cizeau, J.; Jewett, M.A.; MacDonald, G.C. A Phase II Study of Oportuzumab Monatox: An Immunotoxin Therapy for Patients with Noninvasive Urothelial Carcinoma In Situ Previously Treated with Bacillus Calmette-Guérin. J. Urol. 2012, 188, 1712–1718. [Google Scholar] [CrossRef]
- Dickstein, R.; Wu, N.; Cowan, B.; Dunshee, C.; Franks, M.; Wolk, F.; Belkoff, L.; Castellucci, S.; Holzbeierlein, J.; Kulkarni, G.; et al. VISTA, phase 3 trial of Vicinium, an EpCAM-targeted Pseudomonas exotoxin, in BCG-unresponsive non-muscle invasive bladder cancer. Ren. Fail. 2018, 4, 3. [Google Scholar]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2017, 171, 540–556. [Google Scholar] [CrossRef] [Green Version]
- Coates, J.T.; Sun, S.; Leshchiner, I.; Thimmiah, N.; Martin, E.E.; McLoughlin, D.; Danysh, B.P.; Slowik, K.; Jacobs, R.A.; Rhrissorrakrai, K.; et al. Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer. Cancer Discov. 2021, 11, 2436–2445. [Google Scholar] [CrossRef]
- Baselga, J.; Phillips, G.D.L.; Verma, S.; Ro, J.; Huober, J.; Guardino, A.E.; Samant, M.K.; Olsen, S.; De Haas, S.L.; Pegram, M.D. Relationship between Tumor Biomarkers and Efficacy in EMILIA, a Phase III Study of Trastuzumab Emtansine in HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 3755–3763. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Ocana, A.; Tannock, I.F. Reversal of ATP-binding cassette drug transporter activity to modulate chemoresistance: Why has it failed to provide clinical benefit? Cancer Metastasis Rev. 2013, 32, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Mayo, M.F.; Jones, G.E.; Doherty, H.; Maloney, E.K.; Erickson, H.K.; Sun, X.; Wilhelm, S.; Ab, O.; et al. Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res. 2010, 70, 2528–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cianfriglia, M. The biology of MDR1-P-glycoprotein (MDR1-Pgp) in designing functional antibody drug conjugates (ADCs): The experience of gemtuzumab ozogamicin. Ann. Ist. Super Sanita 2013, 49, 150–168. [Google Scholar] [PubMed]
- Lambert, J.M.; Chari, R.V.J. Ado-trastuzumab Emtansine (T-DM1): An Antibody–Drug Conjugate (ADC) for HER2-Positive Breast Cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Sabbaghi, M.; Gil-Gómez, G.; Guardia, C.; Servitja, S.; Arpí, O.; García-Alonso, S.; Menendez, S.; Arumi-Uria, M.; Serrano, L.; Salido, M.; et al. Defective Cyclin B1 Induction in Trastuzumab-emtansine (T-DM1) Acquired Resistance in HER2-positive Breast Cancer. Clin. Cancer Res. 2017, 23, 7006–7019. [Google Scholar] [CrossRef] [Green Version]
- Jedema, I.; Barge, R.M.Y.; Van Der Velden, V.H.J.; A Nijmeijer, B.; Van Dongen, J.J.M.; Willemze, R.; Falkenburg, J.H.F. Internalization and cell cycle-dependent killing of leukemic cells by Gemtuzumab Ozogamicin: Rationale for efficacy in CD33-negative malignancies with endocytic capacity. Leukemia 2003, 18, 316–325. [Google Scholar] [CrossRef]
- Kalim, M.; Chen, J.; Wang, S.; Lin, C.; Ullah, S.; Liang, K.; Ding, Q.; Chen, S.; Zhan, J.-B. Intracellular trafficking of new anticancer therapeutics: Antibody–drug conjugates. Drug Des. Dev. Ther. 2017, 11, 2265–2276. [Google Scholar] [CrossRef] [Green Version]
- Sung, M.; Tan, X.; Lu, B.; Golas, J.; Hosselet, C.; Wang, F.; Tylaska, L.; King, L.; Zhou, D.; Dushin, R.; et al. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-DM1). Mol. Cancer Ther. 2017, 17, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.-D.; Sun, G.; Li, J.; Xu, J.; Wang, X. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett. 2019, 452, 66–70. [Google Scholar] [CrossRef]
- Gerber, H.P.; Sapra, P.; Loganzo, F.; May, C. Combining antibody–drug conjugates and immune-mediated cancer therapy: What to expect? Biochem. Pharmacol. 2016, 102, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; Epp, A.; Law, C.L. Abstract 2469: Brentuximab Vedotin-Mediated Immunogenic Cell Death. Cancer Res. 2015, 75, 2469. Available online: https://aacrjournals.org/cancerres/article/75/15_Supplement/2469/601170/Abstract-2469-Brentuximab-vedotin-mediated (accessed on 24 September 2021).
- Cao, A.T.; Law, C.-L.; Gardai, S.J.; Heiser, R.A. Abstract 5588: Brentuximab vedotin-driven immunogenic cell death enhances antitumor immune responses, and is potentiated by PD1 inhibitionin vivo. Immunology 2017, 77, 5588. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Flaig, T.W.; Friedlander, T.W.; Milowsky, M.I.; Srinivas, S.; Petrylak, D.P.; Merchan, J.R.; Bilen, M.A.; Carret, A.-S.; Yuan, N.; et al. Study EV-103: Preliminary durability results of enfortumab vedotin plus pembrolizumab for locally advanced or metastatic urothelial carcinoma. J. Clin. Oncol. 2020, 38, 441. [Google Scholar] [CrossRef]
- Hoimes, C.J.; Bedke, J.; Loriot, Y.; Nishiyama, H.; Fang, X.; Kataria, R.S.; Moreno, B.H.; Galsky, M.D. KEYNOTE-B15/EV-304: Randomized phase 3 study of perioperative enfortumab vedotin plus pembrolizumab versus chemotherapy in cisplatin-eligible patients with muscle-invasive bladder cancer (MIBC). J. Clin. Oncol. 2021, 39, TPS4587. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug Name | Target | Cytotoxic Payload | Study | Trial Phase | Sample Size | ORR (%) | Median Progression Free Survival (mPFS) in Months (95% CI) | Median Overall Survival (mOS) in Months (95% CI) | Median Duration of Response (mDoR) in Months | Adverse Events (G3–G4) |
|---|---|---|---|---|---|---|---|---|---|---|
| Enfortumab Vedotin | Nectin-4 | MMAE | Rosemberg et al. [74] | 1 | 155 | 43 | 5.4 (5.1–6.3) | 12.3 (9.3–15.3) | 7.4 (5.6–9.6) | 34% |
| Rosemberg et al. [Cohort 1] [75] Y. Yu et al. [Cohort 2] [82] | 2 | 125 89 | 44 52 | 5.8 (4.9–7.5) 5.8 (5.03–8.28) | 11.7 (9.1-not reached) 14.7 (10.51–18.20) | 7.6 (0.95–11.30) 6 (2.8–8.3) | 54% 55% | |||
| Powles et al. [76] | 3 | 608 | 40.6 vs. 17.9 | 5.5 vs. 3.7 (HR, 0.62; 0.51–0.75; p < 0.001) | 12.8 vs. 8.9 (HR, 0.70; 0.56–0.89; p = 0.001) | 5.0 (0.5–19.4) | 51% vs. 48% | |||
| Sacituzumab Govitecan | Trop-2 | SN-38 | Bardia et al. [83] | 1/2 | 45 | 28.9 | 6.8 (3.6–9.7) | 16.8 (9.0–21.9) | 12.9 (3.8–22.5) | 59% |
| Tagawa et al. [84] | 2 | 113 | 27 | 5.4 (3.5–7.2) | 10.9 (9.0–13.8) | 7.2 (4.7–8.6) | not evaluable | |||
| Sirtratumab Vedotin | SLITRK-6 | MMAE | Petrylak et al. (interim analysis) [85] | 1 | 51 | 33 | 4 | - | 3.9 | 50% |
| Disitamab Vedotin | HER-2 | MMAE | Sheng [86] | 2 | 43 | 51.2% | 6.9 (5.6–8.9) | 13.9 (9.1–NE) | 6.9 (4.7–10.8) | 58% |
| NCT Number and Study Name | Drug Name | Setting | Phase | Study Characteristics | Recruitment Status |
|---|---|---|---|---|---|
| NCT03288545 EV-103 | Enfortumab vedotin | Metastatic | I/II | Safety and anticancer activity of Enfortumab vedotin (EV) given intravenously as monotherapy and in combination with other anticancer therapies as first line (1L) and second line (2L) treatment for patients with urothelial cancer. The primary goal of the study is to determine the safety, tolerability, and efficacy of Enfortumab vedotin alone and in combination with pembrolizumab and/or chemotherapy. | Recruiting |
| NCT04223856 EV-302 | Enfortumab vedotin + Pembrolizumab vs chemotherapy | Metastatic | III | An Open-label, Randomized, study of Enfortumab Vedotin in Combination with Pembrolizumab Versus Chemotherapy Alone in Previously Untreated Locally Advanced or Metastatic Urothelial Cancer | Recruiting |
| NCT04225117 EV-202 | Enfortumab vedotin | Metastatic | II | An Open-label, Multicenter, Multicohort, to Evaluate Enfortumab Vedotin in Subjects with Previously Treated Locally Advanced or Metastatic Malignant Solid Tumors | Recruiting |
| NCT04960709 VOLGA | Enfortumab vedotin + Durvalumab +/− tremelimumab | Perioperative | III | Randomized, Open-Label, Multicenter Study to Determine the Efficacy and Safety of Durvalumab in Combination with Tremelimumab and Enfortumab Vedotin or Durvalumab in Combination With Enfortumab Vedotin for Perioperative Treatment in Patients Ineligible for Cisplatin Undergoing Radical Cystectomy for Muscle Invasive Bladder Cancer | Recruiting |
| NCT03924895 KEYNOTE-905/EV-303 | Enfortumab vedotin + Pembro vs. Pembro vs. surgery alone | Perioperative | III | A Randomized Study Evaluating Cystectomy with Perioperative Pembrolizumab and Cystectomy with Perioperative Enfortumab Vedotinand Pembrolizumab Versus Cystectomy Alone in Cisplatin-Ineligible Participants with Muscle-Invasive Bladder Cancer | Recruiting |
| NCT04700124 KEYNOTE-B15/EV-304 | Enfortumab vedotin + Pembrolizumab vs. Cisplatin + Gemcitabine | Perioperative | III | A Randomized, Open-label Study to Evaluate Perioperative Enfortumab Vedotin Plus Pembrolizumab (MK-3475) Versus Neoadjuvant Gemcitabine and Cisplatin in Cisplatin-eligible Participants with Muscle-invasive Bladder Cancer | Recruiting |
| NCT04527991 TROPiCS-04 | Sacituzumab govitecan vs. chemotherapy | Metastatic | III | A Randomized Open-Label Study of Sacituzumab govitecan Versus Treatment of Physician’s Choice in Subjects with Metastatic or Locally Advanced Unresectable Urothelial Cancer | Recruiting |
| NCT03547973 TROPHY-U-01 | Sacitizumab govitecan | Metastatic | II | Open Label, Study of Sacituzumab govitecan in Metastatic Urothelial Cancer After Failure of Platinum-Based Regimen or Anti-PD-1/ PD-L1 Based Immunotherapy | Recruiting |
| NCT04482309 DESTINY-PanTumor02 | Trastuzumab deruxtecan | Metastatic | II | Multicenter, Open-label Study to Evaluate the Efficacy and Safety of Trastuzumab Deruxtecan (T-DXd, DS-8201a) for the Treatment of Selected HER2 Expressing Tumors (DESTINY-PanTumor02) | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungaro, A.; Tucci, M.; Audisio, A.; Di Prima, L.; Pisano, C.; Turco, F.; Delcuratolo, M.D.; Di Maio, M.; Scagliotti, G.V.; Buttigliero, C. Antibody-Drug Conjugates in Urothelial Carcinoma: A New Therapeutic Opportunity Moves from Bench to Bedside. Cells 2022, 11, 803. https://doi.org/10.3390/cells11050803
Ungaro A, Tucci M, Audisio A, Di Prima L, Pisano C, Turco F, Delcuratolo MD, Di Maio M, Scagliotti GV, Buttigliero C. Antibody-Drug Conjugates in Urothelial Carcinoma: A New Therapeutic Opportunity Moves from Bench to Bedside. Cells. 2022; 11(5):803. https://doi.org/10.3390/cells11050803
Chicago/Turabian StyleUngaro, Antonio, Marcello Tucci, Alessandro Audisio, Lavinia Di Prima, Chiara Pisano, Fabio Turco, Marco Donatello Delcuratolo, Massimo Di Maio, Giorgio Vittorio Scagliotti, and Consuelo Buttigliero. 2022. "Antibody-Drug Conjugates in Urothelial Carcinoma: A New Therapeutic Opportunity Moves from Bench to Bedside" Cells 11, no. 5: 803. https://doi.org/10.3390/cells11050803