
Complete Chloroplast Genome of an Endangered Species Quercus litseoides, and Its Comparative, Evolutionary, and Phylogenetic Study with Other Quercus Section Cyclobalanopsis Species

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, DNA Extraction, and Sequencing
2.2. Genome Assembly and Annotations
2.3. Repeated Sequence Analysis
2.4. Genome Structure Comparisons and Sequence Divergence Analysis
2.5. Evolutionary Selection Pressure Analysis
2.6. Phylogenetic Analyses
3. Results
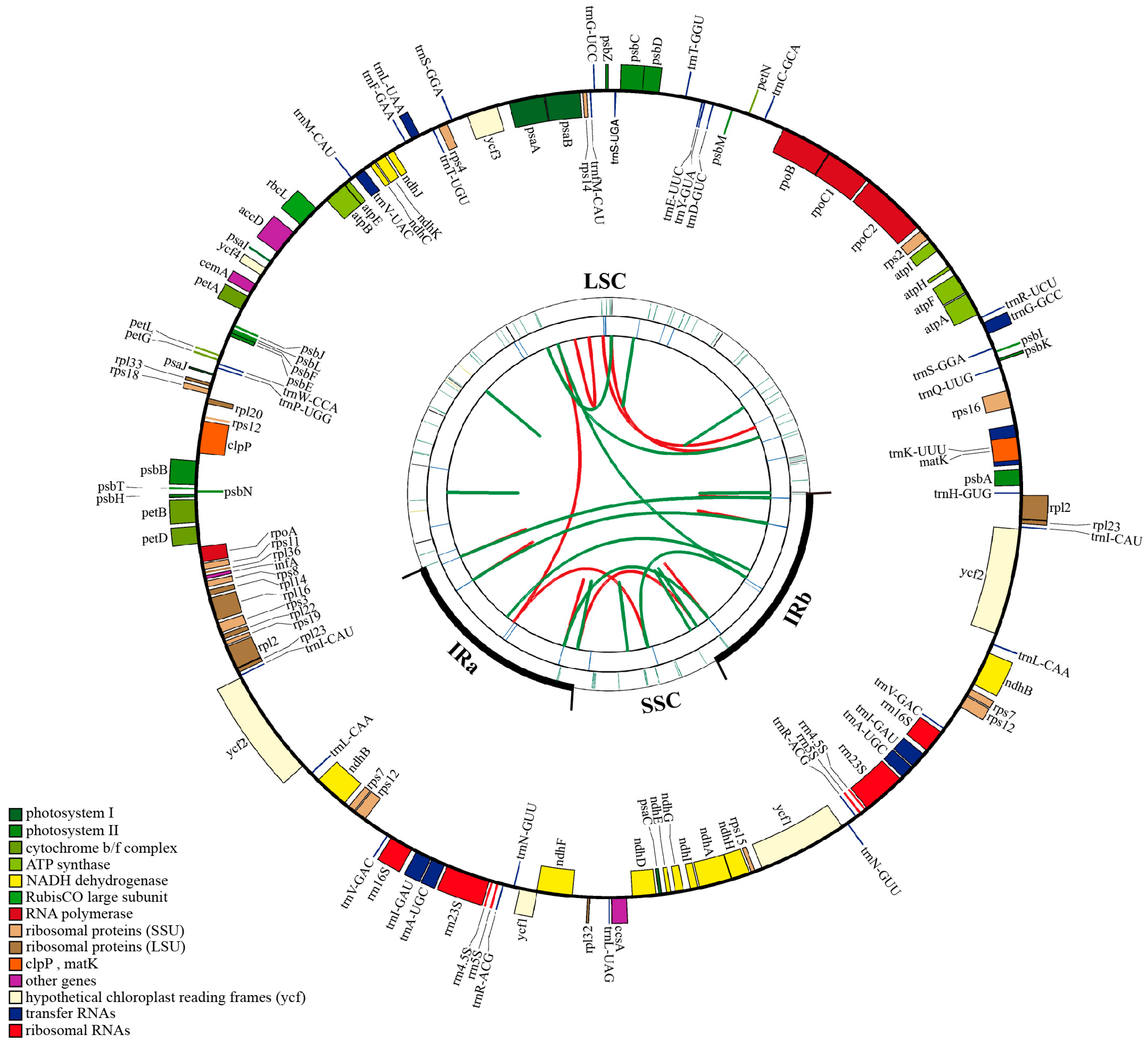
3.1. Chloroplast Genome Assembly and Annotation of Q. litseoides
3.2. Repeat Sequences in the Chloroplast Genome of Q. litseoides
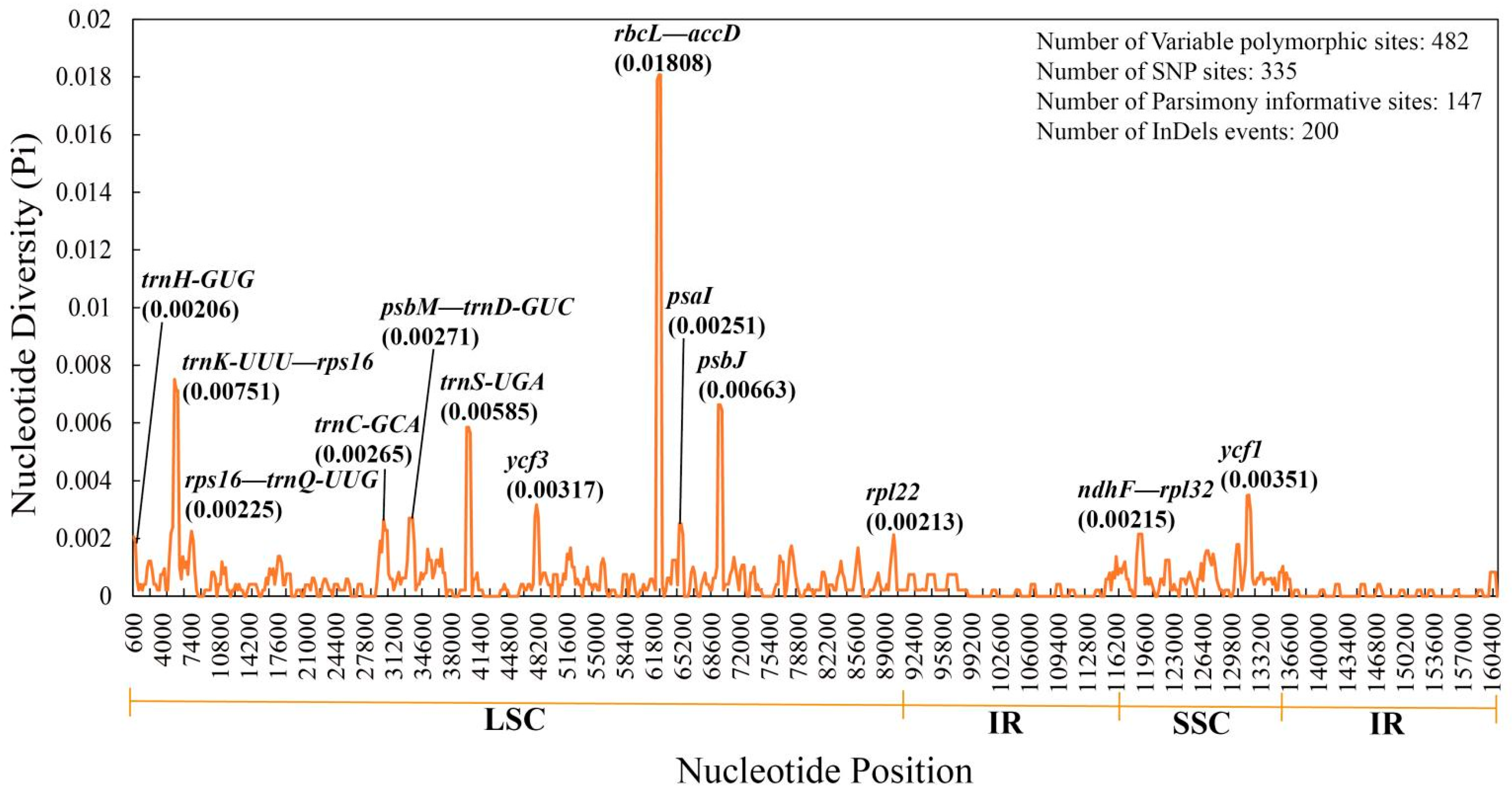
3.3. Genome Structure Comparisons and Sequence Divergence of Quercus Section Cyclobalanopsis
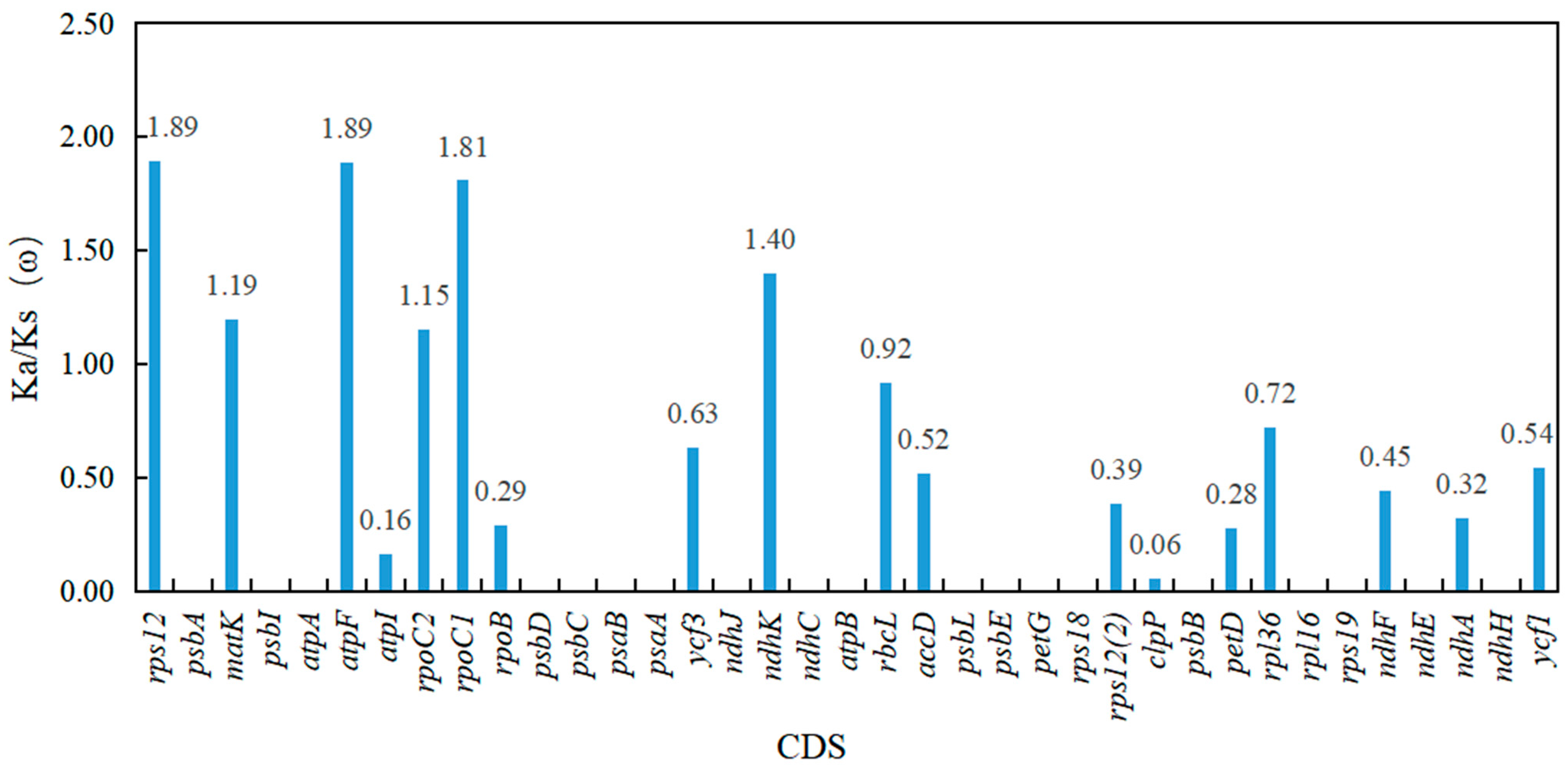
3.4. Selective Pressure Analysis
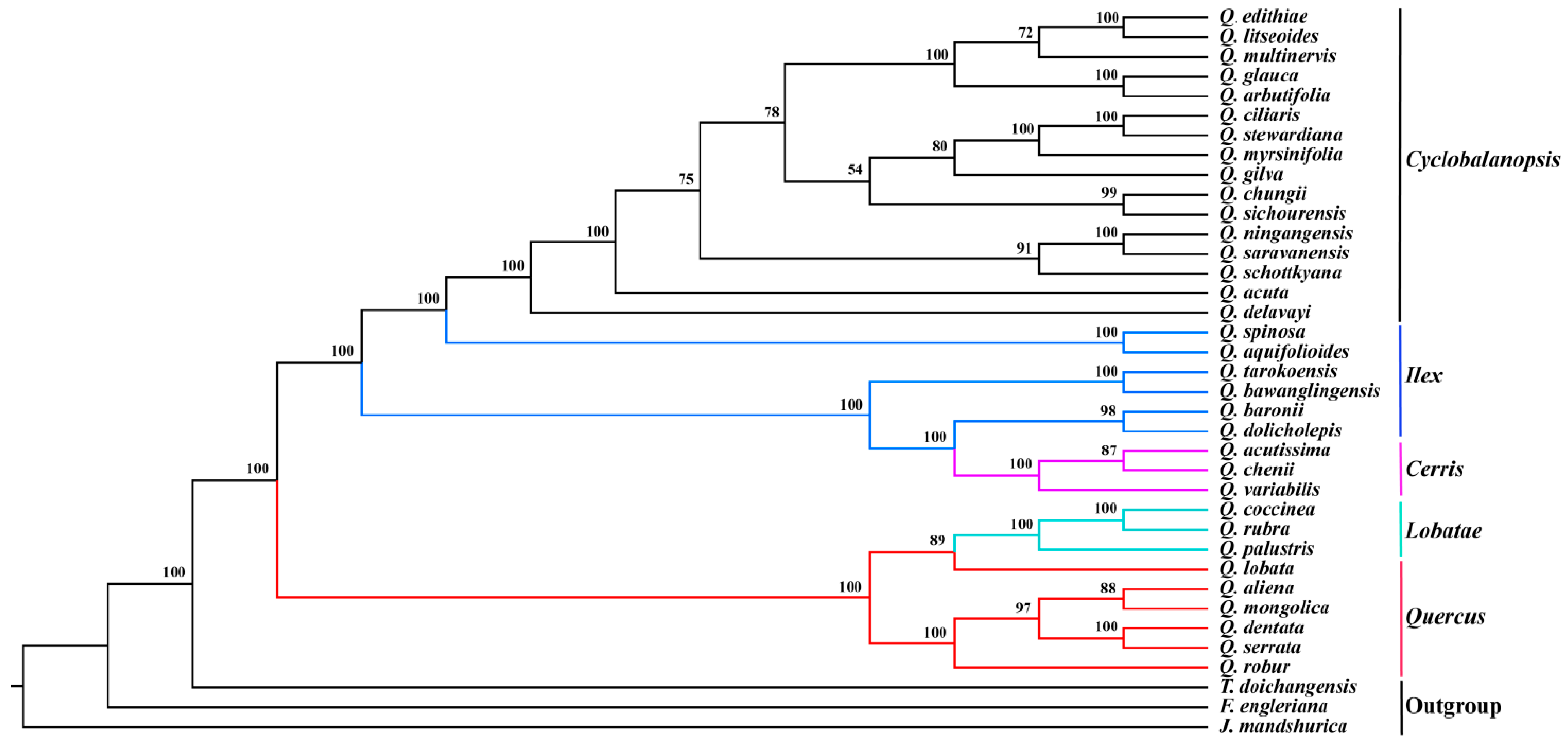
3.5. Phylogenetic Analyses
4. Discussion
4.1. Architecture of cp Genomes in Quercus Section Cyclobalanopsis
4.2. Phylogeny of Chloroplast Genome of Quercus
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- BGCI. State of the World’s Trees; BGCI: Richmond, UK, 2021; pp. 3–6. [Google Scholar]
- Fazan, L.; Song, Y.G.; Kozlowski, G. The Woody Planet: From Past Triumph to Manmade Decline. Plants 2020, 9, 1593. [Google Scholar] [CrossRef]
- Watson, J.E.M.; Evans, T.; Venter, O.; Williams, B.; Tulloch, A.; Stewart, C.; Thompson, I.; Ray, J.C.; Murray, K.; Salazar, A.; et al. The exceptional value of intact forest ecosystems. Nat. Ecol. Evol. 2018, 2, 599–610. [Google Scholar] [CrossRef]
- Denk, T.; Grimm, G.W.; Manos, P.S.; Deng, M.; Hipp, A.L. An Updated Infrageneric Classification of the Oaks: Review of Previous Taxonomic Schemes and Synthesis of Evolutionary Patterns. In Oaks Physiological Ecology. Exploring the Functional Diversity of Genus Quercus L.; Gil-Pelegrin, E., Peguero-Pina, J., Eds.; Tree Physiology: Springer, Cham, 2017; Volume 7, pp. 13–38. [Google Scholar] [CrossRef]
- Deng, M.; Jiang, X.L.; Hipp, A.L.; Manos, P.S.; Hahn, M. Phylogeny and biogeography of East Asian evergreen oaks (Quercus section Cyclobalanopsis; Fagaceae): Insights into the Cenozoic history of evergreen broad-leaved forests in subtropical Asia. Mol. Phylogenet. Evol. 2018, 119, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Hipp, A.L.; Manos, P.S.; Hahn, M.; Avishai, M.; Bodenes, C.; Cavender-Bares, J.; Crowl, A.A.; Deng, M.; Denk, T.; Fitz-Gibbon, S.; et al. Genomic landscape of the global oak phylogeny. New Phytol. 2020, 226, 1198–1212. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, T.; Qian, Z.; Zhao, G. Phylogenetic relationships in Chinese oaks (Fagaceae, Quercus): Evidence from plastid genome using low-coverage whole genome sequencing. Genomics 2021, 113, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Chang, Y.T.; Bartholomew, B. Fagaceae. In Flora of China, English Version; Science Press and Missouri Botanical Garden Press: Beijing, China; St. Louis, MO, USA, 1999; Volume 4, pp. 380–400. [Google Scholar]
- Carrero, C.; Jerome, D.; Beckman, E.; Byrne, A.; Coombes, A.J.; Deng, M.; González-Rodríguez, A.; Hoang, V.S.; Khoo, E.; Nguyen, N.; et al. The Red List of Oaks 2020; The Morton Arboretum: Lisle, IL, USA, 2020; p. 5. [Google Scholar]
- Manos, P.S.; Doyle, J.J.; Nixon, K.C. Phylogeny, biogeography, and processes of molecular differentiation in Quercus subgenus Quercus (Fagaceae). Mol. Phylogenet. Evol. 1999, 12, 333–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, K.C. Quercus. In Flora of North America North of Mexico; Editorial Committee, Ed.; Oxford University Press: New York, NY, USA, 1997; pp. 445–447. [Google Scholar]
- CFH. 2022. Available online: http://cfh.ac.cn/ (accessed on 29 May 2022).
- Song, Y.G.; Petitpierre, B.; Deng, M.; Wu, J.P.; Kozlowski, G. Predicting climate change impacts on the threatened Quercus arbutifolia in montane cloud forests in southern China and Vietnam: Conservation implications. For. Ecol. Manag. 2019, 444, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Deng, M.; Hipp, A.; Song, Y.G.; Li, Q.S.; Coombes, A.; Cotton, A. Leaf epidermal features of Quercus subgenus Cyclobalanopsis (Fagaceae) and their systematic significance. Bot. J. Linn. Soc. 2014, 176, 224–259. [Google Scholar] [CrossRef] [Green Version]
- Bobik, K.; Burch-Smith, T.M. Chloroplast signaling within, between and beyond cells. Front. Plant Sci. 2015, 6, 781. [Google Scholar] [CrossRef] [Green Version]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asaf, S.; Khan, A.L.; Aaqil Khan, M.; Muhammad Imran, Q.; Kang, S.M.; Al-Hosni, K.; Jeong, E.J.; Lee, K.E.; Lee, I.J. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLoS ONE 2017, 12, e0182281. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W.; Maruyama, T.; Fuerst, P. An Approach to Population and Evolutionary Genetic Theory for Genes in Mitochondria and Chloroplasts, and Some Results. Genetics 1983, 103, 513–527. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Qian, J.; Li, X.; Sun, Z.; Xu, X.; Chen, S. Complete Chloroplast Genome of Medicinal Plant Lonicera japonica: Genome Rearrangement, Intron Gain and Loss, and Implications for Phylogenetic Studies. Molecules 2017, 22, 249. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Batzoglou, S.; Berger, B.; Mesirov, J.; Lander, E.S. Sequencing a genome by walking with clone-end sequences: A mathematical analysis. Genome Res. 1999, 9, 1163–1174. [Google Scholar] [CrossRef] [Green Version]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration- supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, gix120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW–A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Wang, L.; Lei, J.; Duan, B.; Ma, W.; Xiao, S.; Qi, H.; Wang, Z.; Liu, Y.; Shen, X.; et al. A Comparative Analysis of the Chloroplast Genomes of Four Salvia Medicinal Plants. Engineering 2019, 5, 907–915. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19 (Suppl. S1), i54–i62. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Tippmann, H.F. Analysis for free: Comparing programs for sequence analysis. Brief Bioinform. 2004, 5, 82–87. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Gou, W.; Jia, S.B.; Price, M.; Guo, X.L.; Zhou, S.D.; He, X.J. Complete Plastid Genome Sequencing of Eight Species from Hansenia, Haplosphaera and Sinodielsia (Apiaceae): Comparative Analyses and Phylogenetic Implications. Plants 2020, 9, 1523–1539. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486. [Google Scholar] [CrossRef]
- Bi, G.; Mao, Y.; Xing, Q.; Cao, M. HomBlocks: A multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching. Genomics 2018, 110, 18–22. [Google Scholar] [CrossRef]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.D. Comparative organization of chloroplast genomes. Annu. Rev. Genet. 1985, 19, 325–354. [Google Scholar] [CrossRef]
- Alexander, L.W.; Woeste, K.E. Pyrosequencing of the northern red oak (Quercus rubra L.) chloroplast genome reveals high quality polymorphisms for population management. Tree Genet. Genomes 2014, 10, 803–812. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Chen, H.; Qi, M.; Su, W.; Zhang, Y.; Du, F.K. The complete chloroplast genome of Quercus fabri (Fagaceae) from China. Mitochondrial DNA Part B 2019, 4, 2857–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Depamphilis, C.W.; Leebens-Mack, J.; Muller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaud, M.F.; Zhang, D.X.; Cerff, R. Differential intron loss and endosymbiotic transfer of chloroplast glyceraldehyde-3- phosphate dehydrogenase genes to the nucleus. Proc. Natl. Acad. Sci. USA 1990, 87, 8918–8922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.Z.; Liu, Y.L.; Zhang, D.; Li, W.; Gao, J.; Liu, Y.; Li, K.; Shi, C.; Zhao, Y.; Zhao, Y.J.; et al. Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2019, 2, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Li, X.; Chen, X.; Huang, X.; Jin, S. The Complete Chloroplast Genome of Carya cathayensis and Phylogenetic Analysis. Genes 2022, 13, 369. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Ji, C.; Chen, Z.; Cai, H.; Wu, X.; Shi, C.; Wang, S. Comparative Analysis the Complete Chloroplast Genomes of Nine Musa Species: Genomic Features, Comparative Analysis, and Phylogenetic Implications. Front. Plant Sci. 2022, 13, 832884. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Wu, X.; Li, T.; Jia, M.; Liu, X.; Liao, L. The complete chloroplast genome of Stauntonia chinensis and compared analysis revealed adaptive evolution of subfamily Lardizabaloideae species in China. BMC Genom. 2021, 22, 161. [Google Scholar] [CrossRef]
- Timme, R.E.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. A comparative analysis of the Lactuca and Helianthus (Asteraceae) plastid genomes: Identification of divergent regions and categorization of shared repeats. Am. J. Bot. 2007, 94, 302–312. [Google Scholar] [CrossRef]
- Weng, M.L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 2014, 31, 645–659. [Google Scholar] [CrossRef] [Green Version]
- Morton, B.R. The influence of neighboring base composition on substitutions in plant chloroplast coding sequences. Mol. Biol. Evol. 1997, 14, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Morton, B.R.; Clegg, M.T. Neighboring Base Composition Is Strongly Correlated with Base Substitution Bias in a Region of the Chloroplast Genome. J. Mol. Evol. 1995, 41, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.S.; Yang, J.; Hu, H.L.; Xia, R.X.; Li, Y.P.; Su, J.F.; Li, Q.; Liu, Y.Q.; Qin, L. A high level of chloroplast genome sequence variability in the Sawtooth Oak Quercus acutissima. Int. J. Biol. Macromol. 2020, 152, 340–348. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chang, E.; Liu, J.; Jiang, Z. Comparative analysis of the complete chloroplast genomes of six white oaks with high ecological amplitude in China. J. For. Res. 2021, 32, 2203–2218. [Google Scholar] [CrossRef]
- Liu, X.; Chang, E.M.; Liu, J.F.; Huang, Y.N.; Wang, Y.; Yao, N.; Jiang, Z.P. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus bawanglingensis Huang, Li et Xing, a Vulnerable Oak Tree in China. Forests 2019, 10, 587. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.R.; Wang, Z.W.; Song, Y.G.; Kozlowski, G. The complete chloroplast genome sequence of Quercus ningangensis and its phylogenetic implication. Plant Fungal Syst. 2021, 66, 155–165. [Google Scholar] [CrossRef]
- Yang, Y.; Hu, Y.; Ren, T.; Sun, J.; Zhao, G. Remarkably conserved plastid genomes of Quercus group Cerris in China: Comparative and phylogenetic analyses. Nord. J. Bot. 2018, 36, e01921. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative Analysis of the Complete Chloroplast Genomes of Five Quercus Species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [Green Version]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef] [Green Version]
- King, D.G.; Soller, M.; Kashi, S.Y. Evolutionary tuning knobs. Endeavour 1997, 21, 36–40. [Google Scholar] [CrossRef]
- Hansen, D.R.; Dastidar, S.G.; Cai, Z.; Penaflor, C.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 2007, 45, 547–563. [Google Scholar] [CrossRef]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marechal, A.; Brisson, N. Recombination and the maintenance of plant organelle genome stability. New Phytol. 2010, 186, 299–317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.W.; Duan, D.; Ma, X.F.; Jia, Y.; Liu, Z.L.; Zhao, G.F.; Li, Z.H. Efficient Identification of the Forest Tree Species in Aceraceae Using DNA Barcodes. Front. Plant Sci. 2016, 7, 1707. [Google Scholar] [CrossRef] [Green Version]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef]
- Wang, X.; Shi, X.; Chen, S.; Ma, C.; Xu, S. Evolutionary Origin, Gradual Accumulation and Functional Divergence of Heat Shock Factor Gene Family with Plant Evolution. Front. Plant Sci. 2018, 9, 71. [Google Scholar] [CrossRef]
- Wicke, S.; Schaferhoff, B.; dePamphilis, C.W.; Muller, K.F. Disproportional plastome-wide increase of substitution rates and relaxed purifying selection in genes of carnivorous Lentibulariaceae. Mol. Biol. Evol. 2014, 31, 529–545. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000; pp. 385–386. [Google Scholar]
- Li, X.; Li, Y.; Sylvester, S.P.; Zang, M.; El-Kassaby, Y.A.; Fang, Y. Evolutionary patterns of nucleotide substitution rates in plastid genomes of Quercus. Ecol. Evol. 2021, 11, 13401–13414. [Google Scholar] [CrossRef]
- Hao, D.C.; Chen, S.L.; Xiao, P.G. Molecular evolution and positive Darwinian selection of the chloroplast maturase matK. J. Plant Res. 2010, 123, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.N.; Ren, Y.; Zhang, J.Q. Conservation and innovation: Plastome evolution during rapid radiation of Rhodiola on the Qinghai-Tibetan Plateau. Mol. Phylogenet. Evol. 2020, 144, 106713. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.F.; Yu, H.X.; Price, M.; Xie, C.; Deng, Y.Q.; Chen, J.P.; Yu, Y.; Zhou, S.D.; He, X.J. Phylogeny of Chinese Allium Species in Section Daghestanica and Adaptive Evolution of Allium (Amaryllidaceae, Allioideae) Species Revealed by the Chloroplast Complete Genome. Front. Plant Sci. 2019, 10, 460. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Feature | Length (bp)/Numbers | GC Content (%) | |
|---|---|---|---|
| Structure length | Total | 160,782 | 36.9 |
| LSC region | 90,235 | 34.74 | |
| SSC region | 18,867 | 31.13 | |
| IR (a/b) region | 25,840 | 42.77 | |
| Gene numbers of different categories | Genes | 131 | 39.5 |
| PCGs | 86 | 37.88 | |
| tRNA | 37 | 53.2 | |
| rRNA | 8 | 55.49 | |
| Gene numbers of different regions | LSC region | 61 (PCGs) and 22 (tRNA) | No information |
| SSC region | 11 (PCGs) and 1 (tRNA) | No information | |
| IR regions | 14 (PCGs), 14 (tRNA) and 8 (rRNA) | No information | |
| Category | Group | Name |
|---|---|---|
| Transcription and translation | Translational initiation factor | infA |
| Ribosomal RNAs | rrn16S (×2), rrn4.5S (×2), rrn23S (×2), rrn5S (×2) | |
| Transfer RNAs | trnR-UCU, trnfM-CAU, trnD-GUC, trnH-GUG, trnM-CAU, trnE-UUC, trnS-GCU, trnF-GAA, trnP-UGG, trnT-UGU, trnG-UCC, trnQ-UUG, trnY-GUA, trnW-CCA, trnS-UGA, trnC-GCA, trnT-GGU, trnL-UAG, trnS-GGA, trnK-UUU *, trnV-UAC *, trnL-UAA *, trnG-GCC *, trnA-UGC *(×2), trnI-GAU *(×2), trnL-CAA (×2), trnI-CAU (×2), trnN-GUU (×2), trnV-GAC (×2), trnR-ACG (×2) | |
| Small subunit of ribosome (SSU) | rps2, rps11, rps19, rps14, rps4, rps15, rps16*, rps8, rps18, rps3, rps12 **(×2), rps7 (×2) | |
| Large subunit of ribosome (LSU) | rpl14, rpl20, rpl36, rpl33, rpl16 *, rpl32, rpl22, rpl2 *(×2), rpl23 (×2) | |
| DNA-dependent RNA polymerase | rpoC2, rpoB, rpoC1 *, rpoA | |
| Photosynthesis | Photosystem I | psaB, psaJ, psaA, psaI, psaC |
| Photosystem II | psbA, psbC, psbH, psbZ, psbI, psbJ, psbK, psbF, psbD, psbT, psbN, psbL, psbM, psbE, psbB | |
| Subunit of cytochrome | petB *, petN, petL, petG, petD *, petA | |
| ATP synthase | atpA, atpI, atpB, atpE, atpF *, atpH | |
| RubisCO large subunit | rbcL | |
| NADH dehydrogenase | ndhG, ndhD, ndhE, ndhK, ndhH, ndhI, ndhF, ndhA *, ndhJ, ndhC, ndhB *(×2) | |
| Biosynthesis | Maturase | matK |
| ATP-dependent Protease | clpP ** | |
| Acetyl-CoA-carboxylase | accD | |
| Envelop membrane protein | cemA | |
| C-Type cytochrome synthesis | ccsA | |
| Unknown | Hypothetical chloroplast reading frames(ycf) | ycf4, ycf3 **, ycf1 (×2), ycf2 (×2) |
| Repeat Type | Repeat Unit | Number (Proportion) of SSRs | Region | Location | |||
|---|---|---|---|---|---|---|---|
| LSC | SSC | IRs | IGS | GR | |||
| Mononucleotides | A/T | 77 (46.67%) | 60 | 11 | 6 | 59 | 18 |
| C/G | 5 (3.03%) | 5 | 0 | 0 | 3 | 2 | |
| Dinucleotides | AG/CT | 19 (11.52%) | 2 | 1 | 16 | 5 | 14 |
| AT/AT | 43 (26.06%) | 29 | 4 | 10 | 28 | 15 | |
| Trinucleotides | AAG/CTT | 1 (0.61%) | 0 | 1 | 0 | 0 | 1 |
| AAT/ATT | 6 (3.64%) | 4 | 2 | 0 | 3 | 3 | |
| Tetranucleotides | AAAT/ATTT | 8 (4.85%) | 7 | 1 | 0 | 5 | 3 |
| AATG/ATTC | 1 (0.61%) | 1 | 0 | 0 | 1 | 0 | |
| AATT/AATT | 2 (1.21%) | 2 | 0 | 0 | 1 | 1 | |
| Pentanucleotides | AAAAT/ATTTT | 1 (0.61%) | 0 | 1 | 0 | 0 | 1 |
| AATGC/ATTGC | 2 (1.21%) | 0 | 0 | 2 | 2 | 0 | |
| Total | 165 (100%) | 110 (66.7%) | 21 (12.7%) | 34 (20.6%) | 107 (64.8%) | 58 (35.2%) | |
| No. | Repeat Type | Repeat Length (bp) | Region | Location | No. | Repeat Type | Repeat Length (bp) | Region | Location |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 19 | LSC | IGS (trnF-GAA, ndhJ) | 25 | R | 33 | LSC, LSC | clpP |
| 2 | M | 20 | IRa | rpl12 | 26 | C | 34 | LSC, LSC | IGS (rps16, trnQ-UUG) |
| 3 | M | 21 | IRa | ycf2 | 27 | C | 30 | LSC, LSC | IGS (petA, psbJ) |
| 4 | M | 31 | IRa | IGS (rrn4.5S, rrn5S) | 28 | P | 56 | SSC, SSC | IGS (ndhD, psaC) |
| 5 | M | 31 | IRb | IGS (rrn5S, rrn4.5S) | 29 | P | 44 | LSC, LSC | IGS (psbT, psbN) |
| 6 | M | 21 | IRb | ycf2 | 30 | P | 40 | IRa, IRb | rpl12 |
| 7 | M | 20 | IRb | rpl12 | 31 | P | 40 | IRa, IRb | rpl12 |
| 8 | F | 40 | IRa, IRa | rpl2 | 32 | P | 38 | LSC, LSC | IGS (atpF, atpH) |
| 9 | F | 40 | IRb, IRb | rpl2 | 33 | P | 34 | SSC, SSC | ycf1 |
| 10 | F | 39 | LSC, IRa | ycf3 | 34 | P | 39 | LSC, IRb | ycf3 |
| 11 | F | 40 | IRa, SSC | IGS (rps12, trnV-GAC) | 35 | P | 40 | SSC, IRb | ndhA |
| 12 | F | 30 | IRa, IRa | IGS (rrn4.5S, rrn5S) | 36 | P | 39 | LSC, LSC | IGS (trnT-GGU, psbD) |
| 13 | F | 30 | IRb, IRb | IGS (rrn5S, rrn4.5S) | 37 | P | 30 | LSC, LSC | trnS-GGA |
| 14 | F | 30 | LSC, LSC | psaB | 38 | P | 30 | IRa, IRb | IGS (rrn4.5S, rrn5S) |
| 15 | F | 30 | LSC, IRa | ycf3 | 39 | P | 30 | IRa, IRb | IGS (rrn4.5S, rrn5S) |
| 16 | F | 30 | IRa, SSC | IGS (rps12, trnV-GAC) | 40 | P | 32 | LSC, LSC | IGS (trnH-GUG, psbA) |
| 17 | F | 30 | IRa, IRb | ycf1 | 41 | P | 30 | LSC, IRb | ycf3 |
| 18 | F | 32 | IRa, IRa | ycf2 | 42 | P | 30 | IRa, IRa | ycf1 |
| 19 | F | 32 | IRb, IRb | ycf2 | 43 | P | 30 | SSC, IRb | ndhA |
| 20 | F | 30 | LSC, LSC | trnS-GGA | 44 | P | 30 | IRb, IRb | ycf1 |
| 21 | F | 30 | LSC, LSC | trnG-GCC | 45 | P | 32 | LSC, LSC | IGS (rbcL, accD) |
| 22 | R | 31 | LSC, LSC | IGS (trnR-UCU, atpA) | 46 | P | 32 | IRa, IRb | ycf2 |
| 23 | R | 31 | LSC, LSC | clpP | 47 | P | 32 | IRa, IRb | ycf2 |
| 24 | R | 31 | LSC, LSC | IGS (atpA, atpF) | 48 | P | 30 | LSC, LSC | trnS-UGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Wang, T.-R.; Kozlowski, G.; Liu, M.-H.; Yi, L.-T.; Song, Y.-G. Complete Chloroplast Genome of an Endangered Species Quercus litseoides, and Its Comparative, Evolutionary, and Phylogenetic Study with Other Quercus Section Cyclobalanopsis Species. Genes 2022, 13, 1184. https://doi.org/10.3390/genes13071184
Li Y, Wang T-R, Kozlowski G, Liu M-H, Yi L-T, Song Y-G. Complete Chloroplast Genome of an Endangered Species Quercus litseoides, and Its Comparative, Evolutionary, and Phylogenetic Study with Other Quercus Section Cyclobalanopsis Species. Genes. 2022; 13(7):1184. https://doi.org/10.3390/genes13071184
Chicago/Turabian StyleLi, Yu, Tian-Rui Wang, Gregor Kozlowski, Mei-Hua Liu, Li-Ta Yi, and Yi-Gang Song. 2022. "Complete Chloroplast Genome of an Endangered Species Quercus litseoides, and Its Comparative, Evolutionary, and Phylogenetic Study with Other Quercus Section Cyclobalanopsis Species" Genes 13, no. 7: 1184. https://doi.org/10.3390/genes13071184