
Complete Chloroplast Genome Sequence of Fagus longipetiolata Seemen (Fagaceae): Genome Structure, Adaptive Evolution, and Phylogenetic Relationships

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction and Genome Sequencing
2.2. Initial Assembly and Annotation of the cp Genome
2.3. Codon Usage and Repeat Sequence Analysis
2.4. Genome Comparison
2.5. Adaptive Evolution and Phylogenetic Analyses
3. Results
3.1. Features of the F. longipetiolata Chloroplast Genome
3.2. Codon Usage Bias
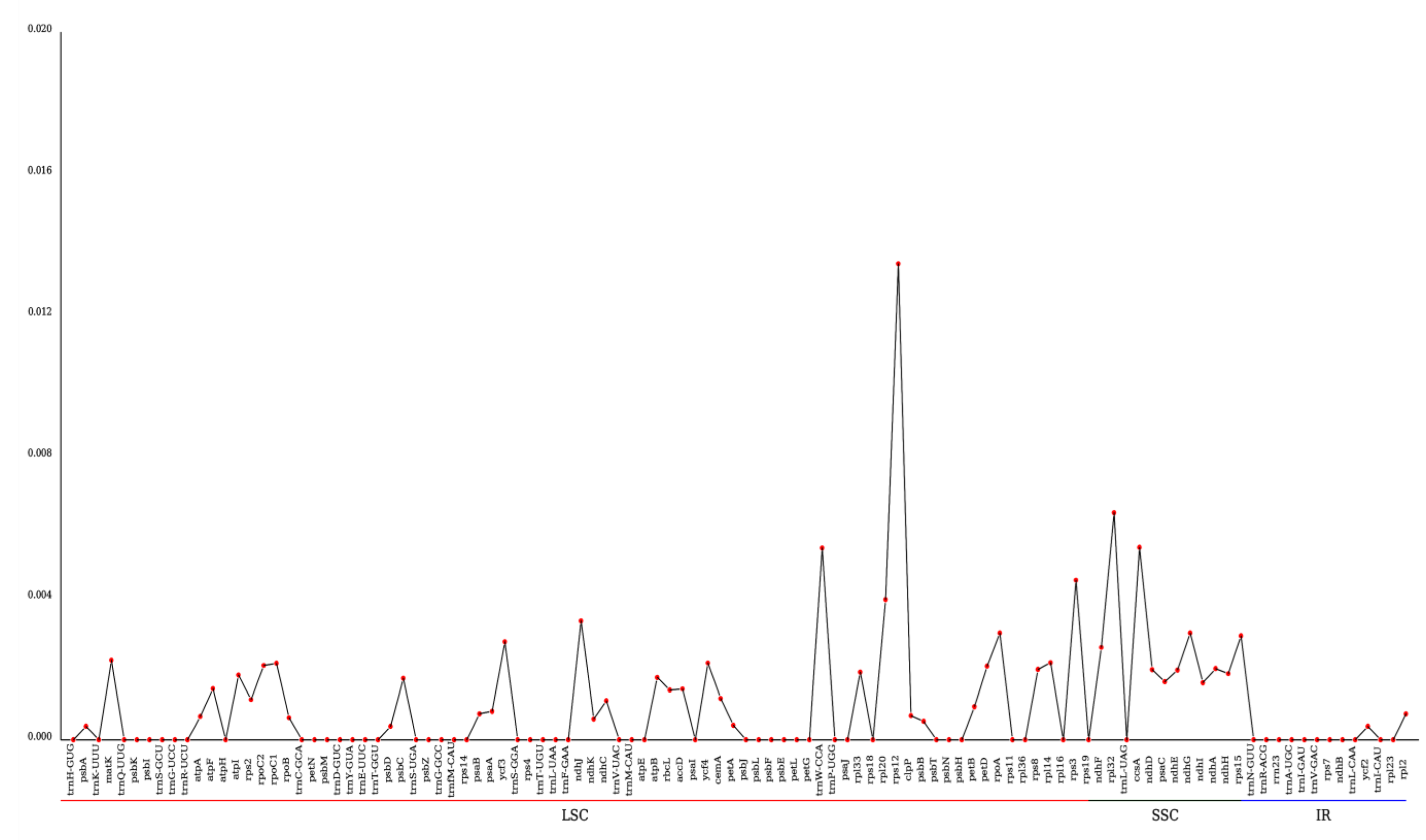
3.3. Detection of Chloroplast Repeat Sequences and SSRs
3.4. Comparison of Complete Chloroplast Genomes
3.5. IR Expansion and Contraction
3.6. Adaptive Evolution Analysis
3.7. Phylogenetic Inference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Renner, S.S.; Grimm, G.W.; Kapli, P.; Denk, T. Species relationships and divergence times in beeches: New insights from the inclusion of 53 young and old fossils in a birth–death clock model. Philos. Trans. R. Soc. B 2016, 371, 20150135. [Google Scholar] [CrossRef] [Green Version]
- Guo, K.; Werger, M.-J. Effect of prevailing monsoons on the distribution of beeches in continental East Asia. For. Ecol. Manag. 2010, 259, 2197–2203. [Google Scholar] [CrossRef]
- Peters, R. Ecology of Beech Forests in the Northern Hemisphere. Ph.D. Thesis, Wageningen Agricultural University, Wageningen, The Netherlands, 1992. [Google Scholar]
- Fang, J.; Lechowicz, M.-J. Climatic limits for the present distribution of beech (Fagus L.) species in the world. J. Biogeogr. 2006, 33, 1804–1819. [Google Scholar] [CrossRef]
- Denk, T. Phylogeny of Fagus L. (Fagaceae) based on morphological data. Plant Syst. Evol. 2003, 240, 55–81. [Google Scholar] [CrossRef]
- Li, J.-Q. On the phylogeny of the Fagaceae. Acta Phytotaxon. Sin. 1996, 34, 597–609. (In Chinese) [Google Scholar]
- Ying, L.-X.; Zhang, T.-T.; Chiu, C.-A.; Chen, T.-Y.; Luo, S.-J.; Chen, X.-Y.; Shen, Z.-H. The phylogeography of Fagus hayatae (Fagaceae): Genetic isolation among populations. Ecol. Evol. 2016, 6, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Nater, A.; Burri, R.; Kawakami, T.; Smeds, L.; Ellegren, H. Resolving evolutionary relationships in closely related species with whole-genome sequencing data. Syst. Biol. 2015, 64, 1000–1017. [Google Scholar] [CrossRef] [Green Version]
- Ravi, V.; Khurana, J.-P.; Tyagi, A.-K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Sugiura, M. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef]
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. 2020, 42, 1–13. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.-B.; Beck, J.-T.; Farmer, S.-B.; Liu, W.; Miller, J.; Siripun, K.-C.; Winder, C.-T.; Schilling, E.-E.; Small, R.-L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhu, J.; Feng, L.; Zhou, T.; Bai, G.; Yang, J.; Zhao, G. Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: Highlighting the effect of codon composition bias in phylogenetic inference. Plant Sci. 2018, 9, 82. [Google Scholar] [CrossRef]
- Worth, J.-R.; Liu, L.; Wei, F.-J.; Tomaru, N. The complete chloroplast genome of Fagus crenata (subgenus Fagus) and comparison with F. engleriana (subgenus Engleriana). Peer J. 2019, 7, e7026. [Google Scholar] [CrossRef]
- Mader, M.; Liesebach, H.; Liesebach, M.; Kersten, B. The complete chloroplast genome sequence of Fagus sylvatica L. (Fagaceae). Mitochondrial DNA Part B 2019, 4, 1818–1819. [Google Scholar] [CrossRef]
- Park, J.; Oh, S.-H. A second complete chloroplast genome sequence of Fagus multinervis Nakai (Fagaceae): Intraspecific variations on chloroplast genome. Mitochondrial DNA Part B 2020, 5, 1868–1869. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Wang, Z.; Tang, Z. Atlas of Woody Plants in China: Distribution and Climate; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011; p. 162. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.-A.; Dvorkin, M.; Kulikov, A.-S.; Pevzner, P.-A. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Takayama, K.; Youn, J.-S.; Pak, J.-H.; Kim, S.-C. Plastome characterization and phylogenomics of East Asian beeches with a special emphasis on Fagus multinervis on Ulleung Island, Korea. Genes 2020, 11, 1338. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.-K.; Jansen, R.-K.; Boore, J.-L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.-H.; Smith, D.-K.; Rabadan, R.; Peiris, M.; Poon, L.-L. Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol. Biol. 2010, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stothard, P.; Grant, J.R.; Van Domselaar, G. Visualizing and comparing circular genomes using the CGView family of tools. Brief. Bioinf. 2019, 20, 1576–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.-C.; Mau, B.; Blattner, F.-R.; Perna, N.-T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.-M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Alberti, S. The origin of the genetic code and protein synthesis. J. Mol. Evol. 1997, 45, 352–358. [Google Scholar] [CrossRef]
- Grantham, R.; Gautier, C.; Gouy, M.; Mercier, R.; Pave, A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Bertranpetit, J.; Oliver, J.-L.; Medina, J.-R. Variation in G + C-content and codon choice: Differences among synonymous codon groups in vertebrate genes. Nucleic Acids Res. 1989, 17, 6181–6189. [Google Scholar] [CrossRef] [Green Version]
- Duret, L.; Mouchiroud, D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 4482–4487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Zhou, T.; Ma, J.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res. 2004, 101, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Van der Linden, M.-G.; de Farias, S.-T. Correlation between codon usage and thermostability. Extremophiles 2006, 10, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Woeste, K.-E.; Zhao, P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 2017, 7, 1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.-Y.; Gao, L.-Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raubeson, L.-A.; Peery, R.; Chumley, T.-W.; Dziubek, C.; Fourcade, H.-M.; Boore, J.-L.; Jansen, R.-K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Ma, L.; Wu, Z.; Chen, K.; Wang, Y. Comparative analyses of chloroplast genomes from 22 Lythraceae species: Inferences for phylogenetic relationships and genome evolution within Myrtales. BMC Plant Biol. 2019, 19, 281. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Lee, J.W.; Choi, B.K. Seven Complete Chloroplast Genomes from Symplocos: Genome Organization and Comparative Analysis. Forests 2021, 12, 608. [Google Scholar] [CrossRef]
- Liu, X.; Chang, E.-M.; Liu, J.-F.; Huang, Y.-N.; Wang, Y.; Yao, N.; Jiang, Z.-P. Complete chloroplast genome sequence and phylogenetic analysis of Quercus bawanglingensis Huang, Li et Xing, a vulnerable oak tree in China. Forests 2019, 10, 587. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Chen, J. Comparison and Phylogenetic Analyses of Nine Complete Chloroplast Genomes of Zingibereae. Forests 2021, 12, 710. [Google Scholar] [CrossRef]
- Ding, S.; Dong, X.; Yang, J.; Guo, C.; Cao, B.; Guo, Y.; Hu, G. Complete chloroplast genome of Clethra fargesii Franch., an original sympetalous plant from central China: Comparative analysis, adaptive evolution, and phylogenetic relationships. Forests 2021, 12, 441. [Google Scholar] [CrossRef]
- Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.-T. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): Comparative analyses and identification of mutational hotspots. Genomics 2020, 112, 581–591. [Google Scholar]
- Yang, Y.; Hu, Y.; Ren, T.; Sun, J.; Zhao, G. Remarkably conserved plastid genomes of Quercus group Cerris in China: Comparative and phylogenetic analyses. Nord. J. Bot. 2018, 36, e01921. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hu, Y.; Liu, M.; Lang, T. Optimization of assembly pipeline may improve the sequence of the chloroplast genome in Quercus spinosa. Sci. Rep. 2018, 8, 8906. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Xu, C.; Liu, Y.; Shi, J.; Li, W.; Suo, Z. Chloroplast phylogenomics and divergence times of Lagerstroemia (Lythraceae). BMC Genom. 2021, 22, 434. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.Q.; Nguyen, T.N.L.; Doan, T.N.; Nguyen, T.T.N.; Phạm, M.H.; Le, T.L.; Sy, T.D.; Chu, H.H.; Chu, H.M. Complete chloroplast genome of novel Adrinandra megaphylla Hu species: Molecular structure, comparative and phylogenetic analysis. Sci. Rep. 2021, 11, 11731. [Google Scholar] [CrossRef]
- Hsu, C.-M.; Yang, W.-P.; Chen, C.-C.; Lai, Y.-K.; Lin, T.-Y. A point mutation in the chloroplast rps12 gene from Nicotiana plumbaginifolia confers streptomycin resistance. Plant Mol. Biol. 1993, 23, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Jansen, R.-K.; Park, S. Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant. Biol. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, D.-K.; Das, A.; Huang, X.; Cianzio, S.; Bhattacharyya, M.-K. Tightly linked Rps12 and Rps13 genes provide broad-spectrum Phytophthora resistance in soybean. Sci. Rep. 2021, 11, 16907. [Google Scholar] [CrossRef]
- Li, W.; Liu, Y.; Yang, Y.; Xie, X.; Lu, Y.; Yang, Z.; Jin, X.; Suo, Z. Interspecific chloroplast genome sequence diversity and genomic resources in Diospyros. BMC Plant Biol. 2018, 18, 210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tian, Y.; Tng, D.-Y.; Zhou, J.; Zhang, Y.; Wang, Z.; Li, P.; Wang, Z. Comparative chloroplast genomics of Litsea Lam. (Lauraceae) and its phylogenetic implications. Forests 2021, 12, 744. [Google Scholar] [CrossRef]
- Hong, Z.; Wu, Z.; Zhao, K.; Yang, Z.; Zhang, N.; Guo, J.; Xu, D. Comparative analyses of five complete chloroplast genomes from the genus Pterocarpus (Fabacaeae). Int. J. Mol. Sci. 2020, 21, 3758. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, A.; Al-Harrasi, A. Unraveling the chloroplast genomes of two prosopis species to identify its genomic information, comparative analyses and phylogenetic relationship. Int. J. Mol. Sci. 2020, 21, 3280. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Sylvester, S.-P.; Li, M.; Zhang, C.; Li, X.; Duan, Y.-F.; Wang, X.-R. The complete plastid genome of Magnolia zenii and genetic comparison to Magnoliaceae species. Molecules 2019, 24, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Long, W.; Li, X. Patterns of synonymous codon usage bias in chloroplast genomes of seed plants. For. Stud. China 2008, 10, 235–242. [Google Scholar] [CrossRef]
- Necşulea, A.; Lobry, J.-R. A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol Evol. 2007, 24, 2169–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orešič, M.; Shalloway, D. Specific correlations between relative synonymous codon usage and protein secondary structure. J. Mol. Biol. 1998, 281, 31–48. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, N.; Wu, H. Analyzing and characterizing the chloroplast genome of Salix wilsonii. BioMed Res. Int. 2019, 2019, 5190425. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Asaf, S.; Khan, A.-L.; Al-Harrasi, A.; Al-Sudairy, O.; AbdulKareem, N.-M.; Shinwari, Z.-K. First complete chloroplast genomics and comparative phylogenetic analysis of Commiphora gileadensis and C. foliacea: Myrrh producing trees. PLoS ONE 2019, 14, e0208511. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zheng, Y. Dynamic evolution and phylogenomic analysis of the chloroplast genome in Schisandraceae. Sci. Rep. 2018, 8, 9285. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Sakaguchi, S.; Isagi, Y.; Setoguchi, H. Comparative chloroplast genomics of series Sakawanum in genus Asarum (Aristolochiaceae) to develop single nucleotide polymorphisms (SNPs) and simple sequence repeat (SSR) markers. J. For. Res. 2018, 23, 387–392. [Google Scholar] [CrossRef]
- Yamane, K.; Kawahara, T. Size homoplasy and mutational behavior of chloroplast simple sequence repeats (cpSSRs) inferred from intra-and interspecific variations in four chloroplast regions of diploid and polyploid Triticum and Aegilops species. Genet. Resour. Crop. Evol. 2018, 65, 727–743. [Google Scholar] [CrossRef]
- Alanazi, K.M.; Ali, M.A.; Kim, S.Y.; Rahman, M.O.; Farah, M.A.; Alhemaid, F.; Elangbam, M.; Gurung, A.B.; Lee, J. The cp genome characterization of Adenium obesum: Gene content, repeat organization and phylogeny. Saudi J. Biol. Sci. 2021, 28, 3768–3775. [Google Scholar] [CrossRef]
- Wu, L.; Cui, Y.; Wang, Q.; Xu, Z.; Wang, Y.; Lin, Y.; Song, J.; Yao, H. Identification and phylogenetic analysis of five Crataegus species (Rosaceae) based on complete chloroplast genomes. Planta 2021, 254, 14. [Google Scholar] [CrossRef]
- Li, D.M.; Zhao, C.Y.; Liu, X.F. Complete Chloroplast Genome Sequences of Kaempferia galanga and Kaempferia elegans: Molecular Structures and Com-parative Analysis. Molecules 2019, 24, 474. [Google Scholar] [CrossRef] [Green Version]
- Mo, Z.; Lou, W.; Chen, Y.; Jia, X.; Zhai, M.; Guo, Z.; Xuan, J. The chloroplast genome of Carya illinoinensis: Genome structure, adaptive evolution, and phylogenetic analysis. Forests 2020, 11, 207. [Google Scholar] [CrossRef] [Green Version]
- Loewe, L.; Charlesworth, B.; Bartolomé, C.; Noel, V. Estimating selection on nonsynonymous mutations. Genetics 2006, 172, 1079–1092. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Lu, P.; Zhang, Z.; Wu, J.-Q.; Zhang, H.; Shen, H. Chloroplast genome sequence of Chongming lima bean (Phaseolus lunatus L.) and comparative analyses with other legume chloroplast genomes. BMC Genom. 2021, 22, 194. [Google Scholar] [CrossRef]
- Xiong, Y.; Xiong, Y.; He, J.; Yu, Q.; Zhao, J.; Lei, X.; Ma, X. The complete chloroplast genome of two important annual clover species, Trifolium alexandrinum and T. resupinatum: Genome structure, comparative analyses and phylogenetic relationships with relatives in Leguminosae. Plants 2020, 9, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Sablok, G.; Wang, B.; Qu, D.; Barbaro, E.; Viola, R.; Varotto, C. Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genom. 2015, 16, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Huang, R.; Li, F.; Tian, E.; Li, C.; Chao, Z. Phylogenetic position of Bupleurum sikangense inferred from the complete chloroplast genome sequence. Gene 2021, 798, 145801. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.M.; Friso, G.; Van Wijk, K.J.; Sloan, D.B. Extreme variation in rates of evolution in the plastid Clp protease complex. Plant J. 2019, 98, 243–259. [Google Scholar] [CrossRef]
- Oh, S.-H.; Manos, P.-S. Molecular phylogenetics and cupule evolution in Fagaceae as inferred from nuclear CRABS CLAW sequences. Taxon 2008, 57, 434–451. [Google Scholar]
- Zhou, Z.-K. Fossils of the Fagaceae and their implications in systematics and biogeography. J. Syst. Evol. 1999, 37, 369–385. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Item | Describe |
|---|---|---|
| Chloroplast genome structure | Cp gene/bp | 158,350 |
| LSC/bp | 87,671 | |
| SSC/bp | 18,891 | |
| IRA/IRB/bp | 25,894 | |
| Gene composition | Cp gene | 131 |
| CDS | 81 | |
| tRNA | 37 | |
| rRNA | 8 | |
| pseudo | 5 | |
| GC Content (%) | Cp gene | 37.09 |
| LSC | 35.05 | |
| SSC | 31.19 | |
| IRA/IRB | 42.70 |
| Category | Gene Group | Gene Name |
|---|---|---|
| Photosynthesis | Subunits of photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Subunits of NADH dehydrogenase | ndhA *, ndhB * (2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Subunits photochlorophyllide reductase | - | |
| Self-replication | Proteins of large ribosomal subunit | # rpl22, rpl14, rpl16 *, rpl2 * (2), rpl20, rpl23 (2), rpl32, rpl33, rpl36 |
| Proteins of small ribosomal subunit | # rps16, rps11, rps12 * (2), rps14, rps15, rps18, rps19, rps2, rps3, rps4, rps7 (2), rps8 | |
| Subunits of RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | |
| Ribosomal RNAs | rrn16 (2), rrn23 (2), rrn4.5 (2), rrn5 (2) | |
| Transfer RNAs | trnA-UGC * (2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnG-UCC *, trnH-GUG, trnI-CAU (2), trnI-GAU * (2), trnK-UUU *, trnL-CAA (2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU (2), trnP-UGG, trnQ-UUG, trnR-ACG (2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC (2), trnV-UAC *, trnW-CCA, trnY-GUA, trnfM-CAU | |
| Other genes | Maturase | matK |
| Protease | clpP ** | |
| Envelope membrane protein | cemA | |
| Acetyl-CoA carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Translation initiation factor | # infA | |
| other | - | |
| Genes of unknown function | Conserved hypothetical chloroplast ORF | # ycf1 (2), ycf2 (2), ycf3 **, ycf4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, D.; Wang, H.; Zhang, J.; Zhao, Y.; Wu, F. Complete Chloroplast Genome Sequence of Fagus longipetiolata Seemen (Fagaceae): Genome Structure, Adaptive Evolution, and Phylogenetic Relationships. Life 2022, 12, 92. https://doi.org/10.3390/life12010092
Liang D, Wang H, Zhang J, Zhao Y, Wu F. Complete Chloroplast Genome Sequence of Fagus longipetiolata Seemen (Fagaceae): Genome Structure, Adaptive Evolution, and Phylogenetic Relationships. Life. 2022; 12(1):92. https://doi.org/10.3390/life12010092
Chicago/Turabian StyleLiang, Daqu, Haoyun Wang, Jun Zhang, Yuanxiang Zhao, and Feng Wu. 2022. "Complete Chloroplast Genome Sequence of Fagus longipetiolata Seemen (Fagaceae): Genome Structure, Adaptive Evolution, and Phylogenetic Relationships" Life 12, no. 1: 92. https://doi.org/10.3390/life12010092