
Chloroplast Genome of Lithocarpus dealbatus (Hook.f. & Thomson ex Miq.) Rehder Establishes Monophyletic Origin of the Species and Reveals Mutational Hotspots with Taxon Delimitation Potential

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Isolation and Sequencing on MGI Platform
2.2. Chloroplast-Genome Assembly
2.3. Annotation and Map Drawing of Chloroplast Genome
2.4. Comparative Analysis of L. dealbatus with Fagaceae Chloroplast Genome for Structural Rearrangement, Similarity, Expansion and Contraction of IR and Tandem Repeat
2.5. DNA Diversity and Ka/Ks Analysis in Lithocarpus
2.6. Phylogenetic Analysis and Estimation of the Divergence Time
3. Results
3.1. Assembly of Chloroplast Genome and Annotation
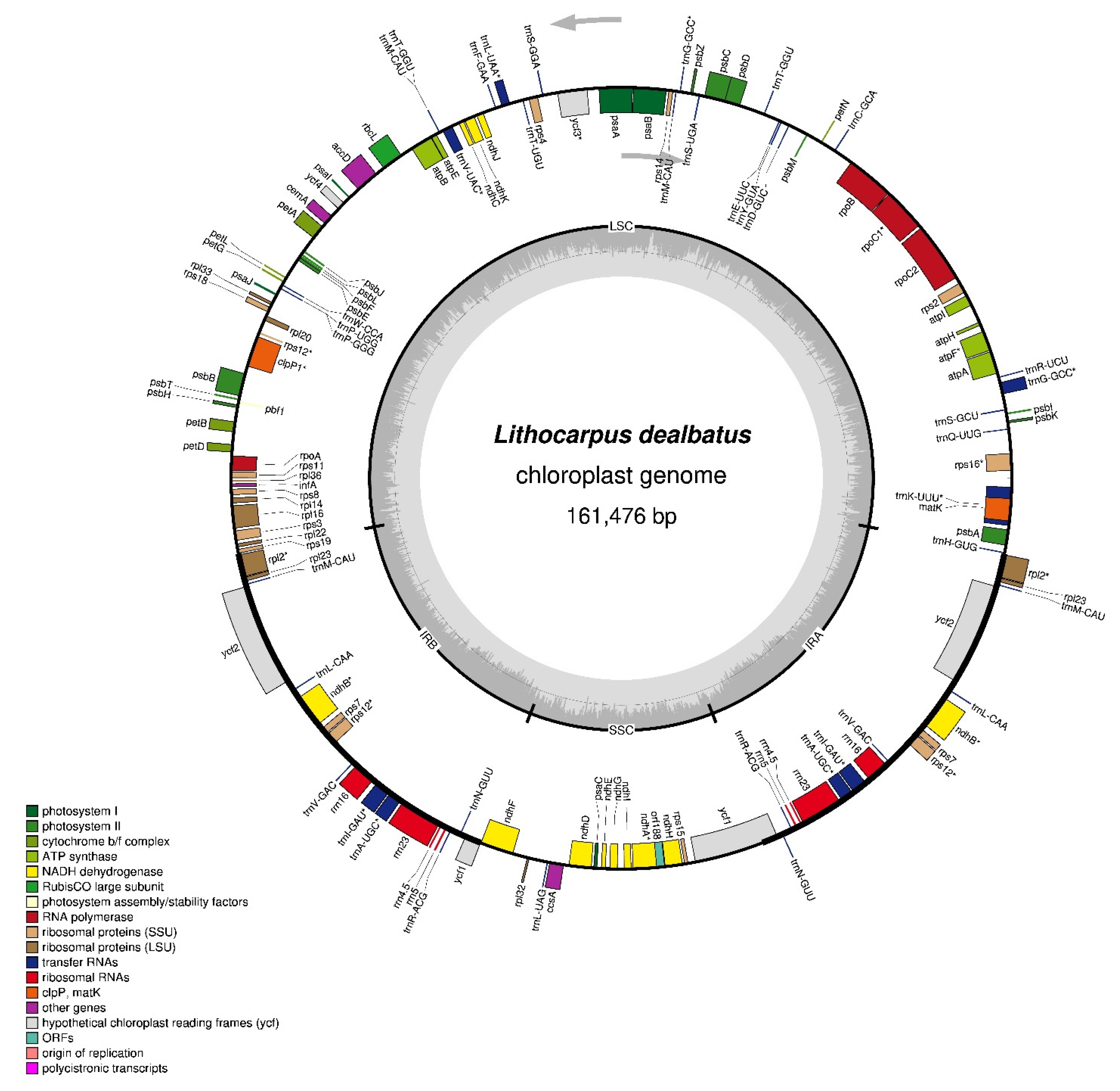
3.1.1. L. dealbatus Chloroplast-Genome Assembly and Architecture
3.1.2. Chloroplast-Genome-Encoding Genes
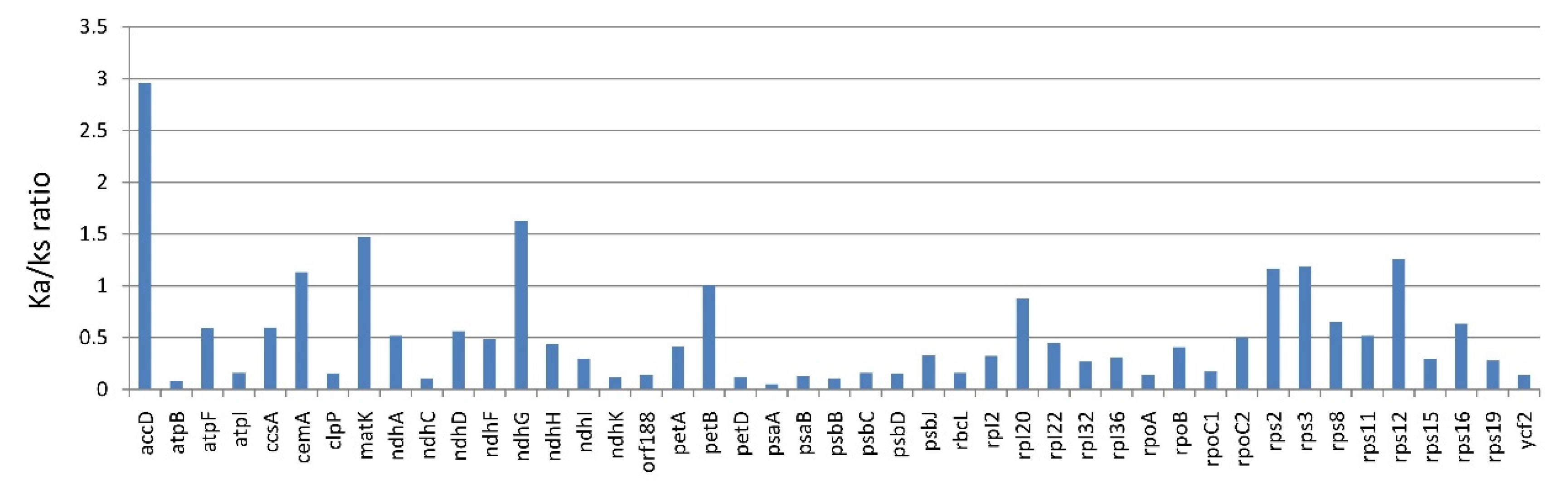
3.1.3. Substitution Rate of Protein-Coding Genes of Lithocarpus
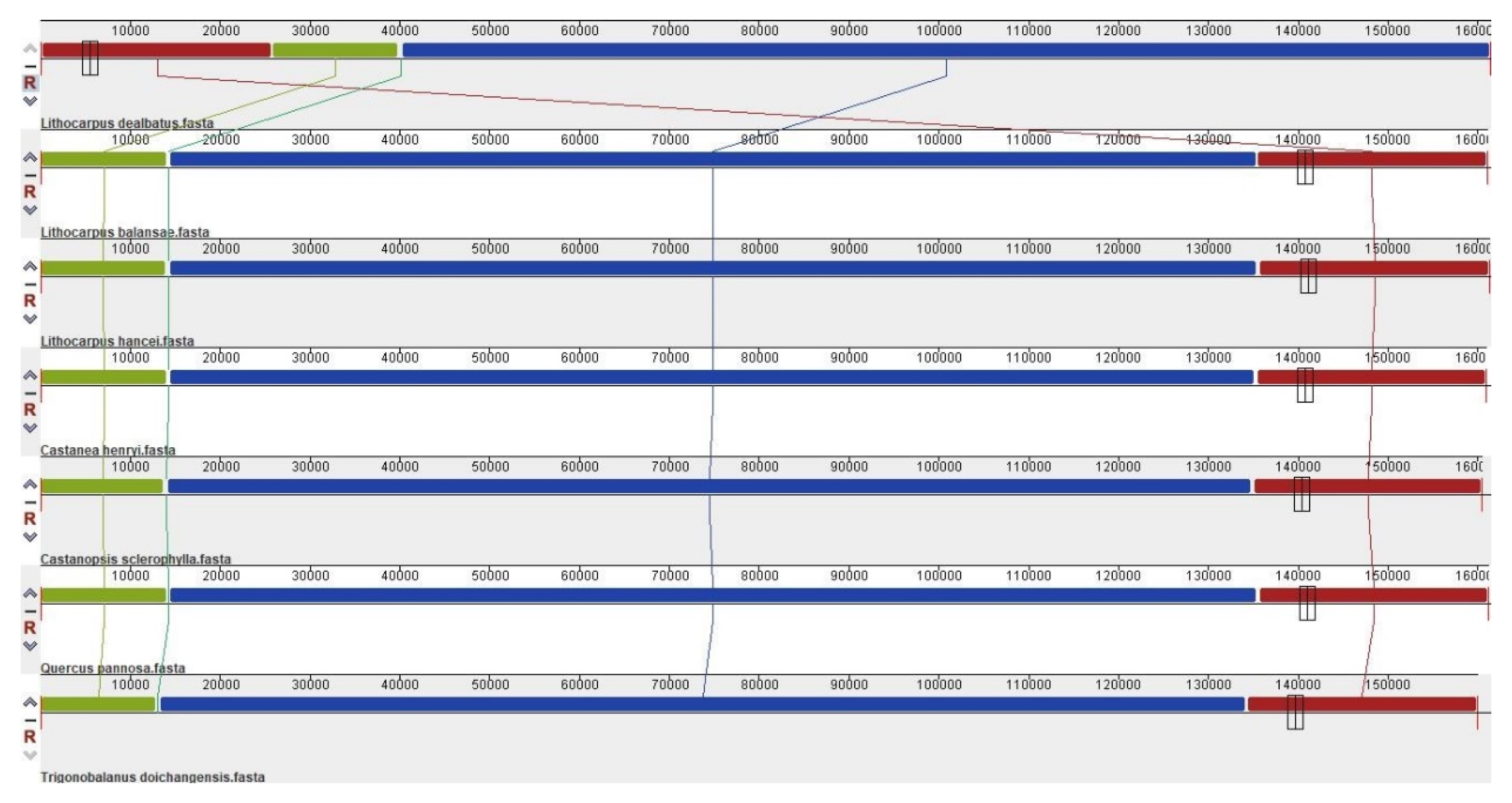
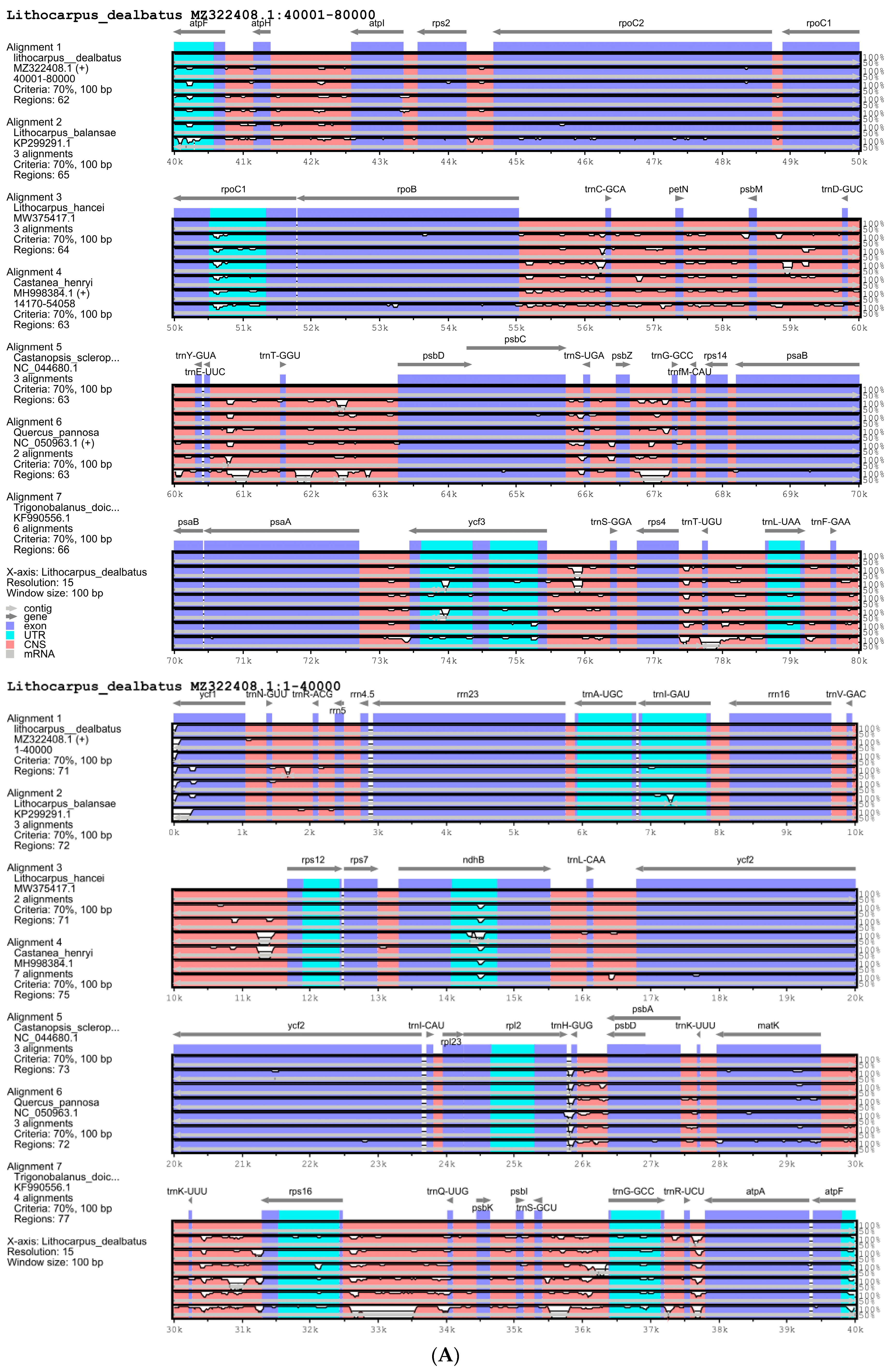
3.2. Comparative Chloroplast Genomes in Quercoideae
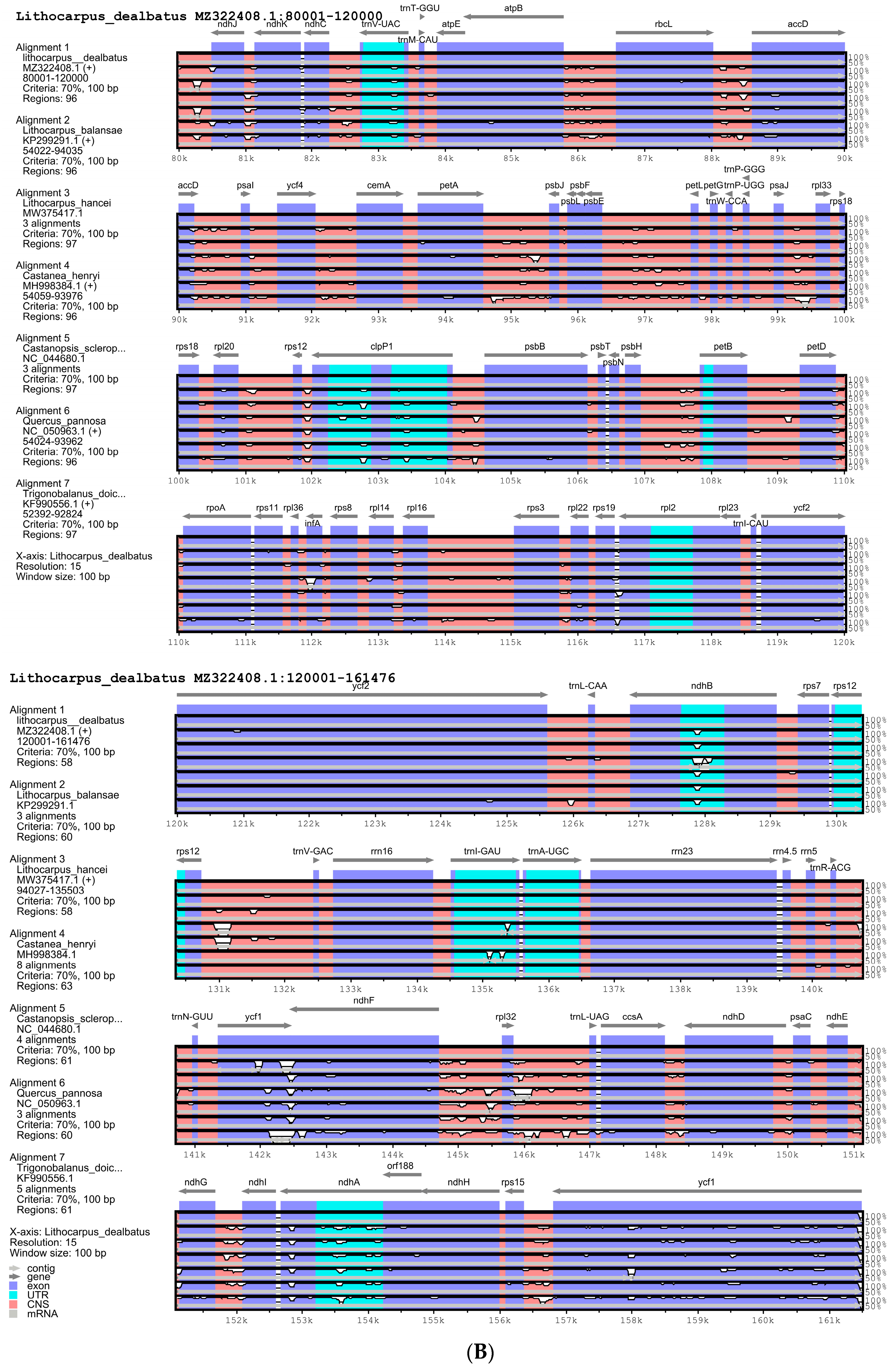
3.2.1. Comparison of Quercoideae Chloroplast Genomes
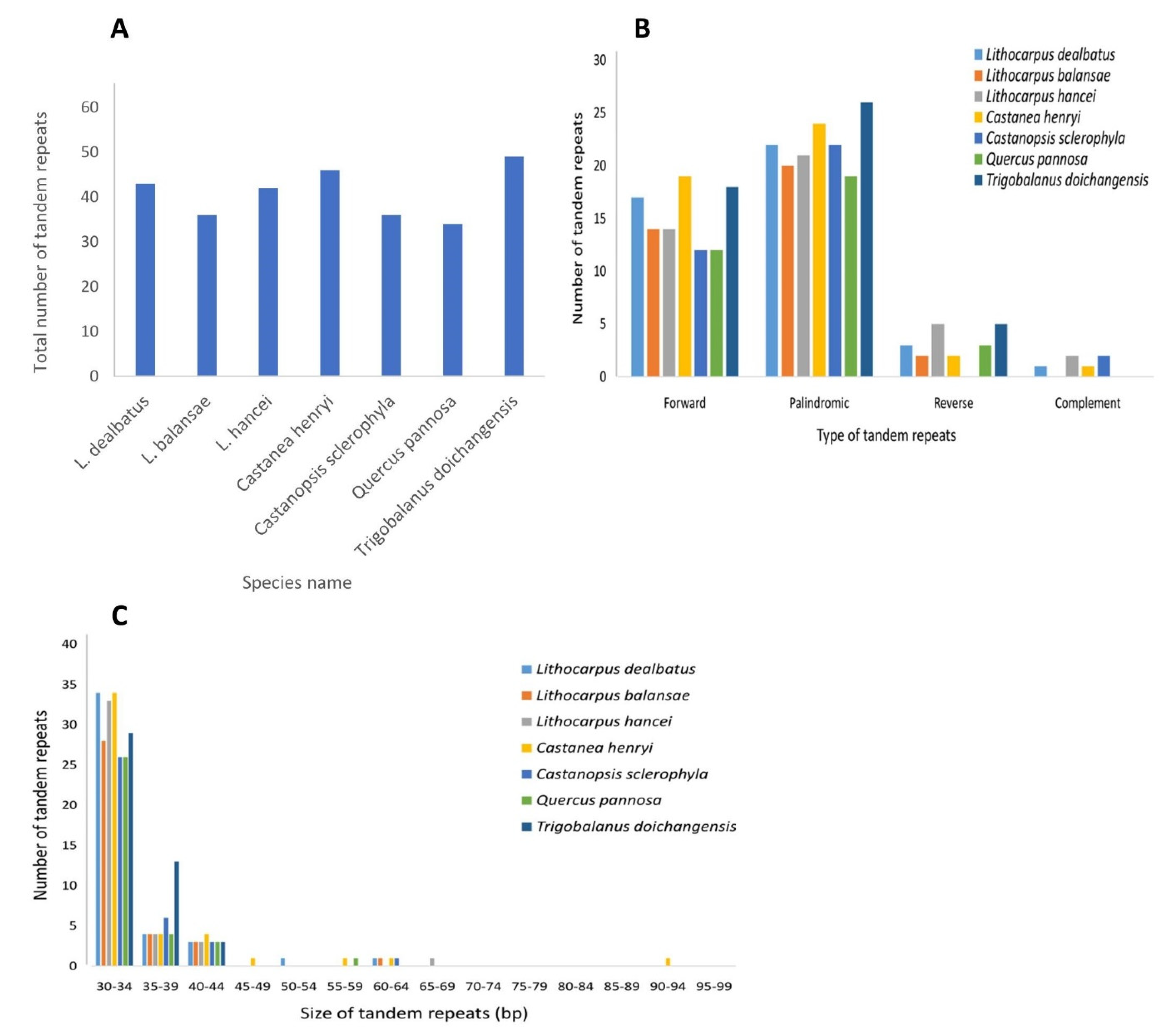
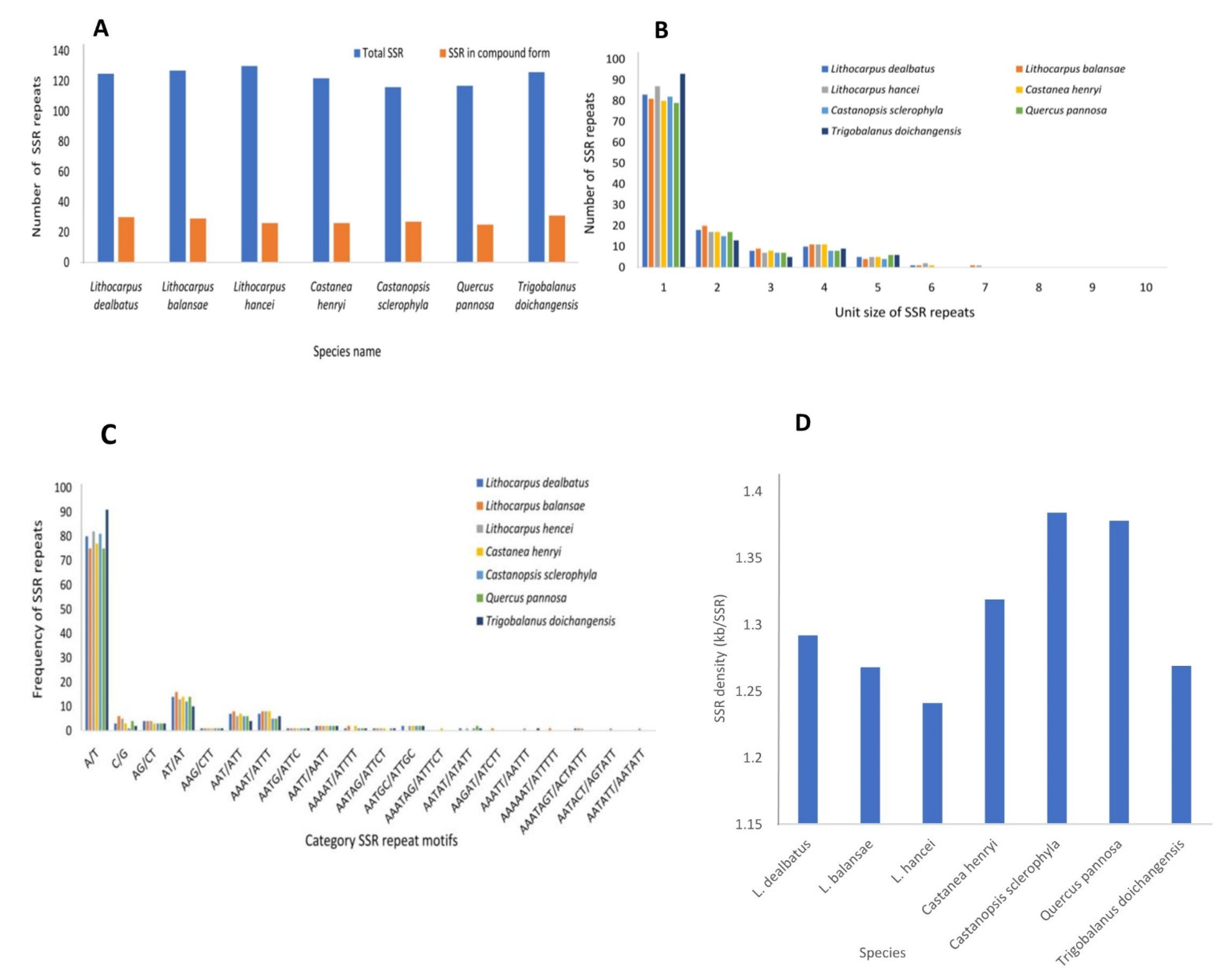
3.2.2. Repeat Sequences and Its Comparative Analysis
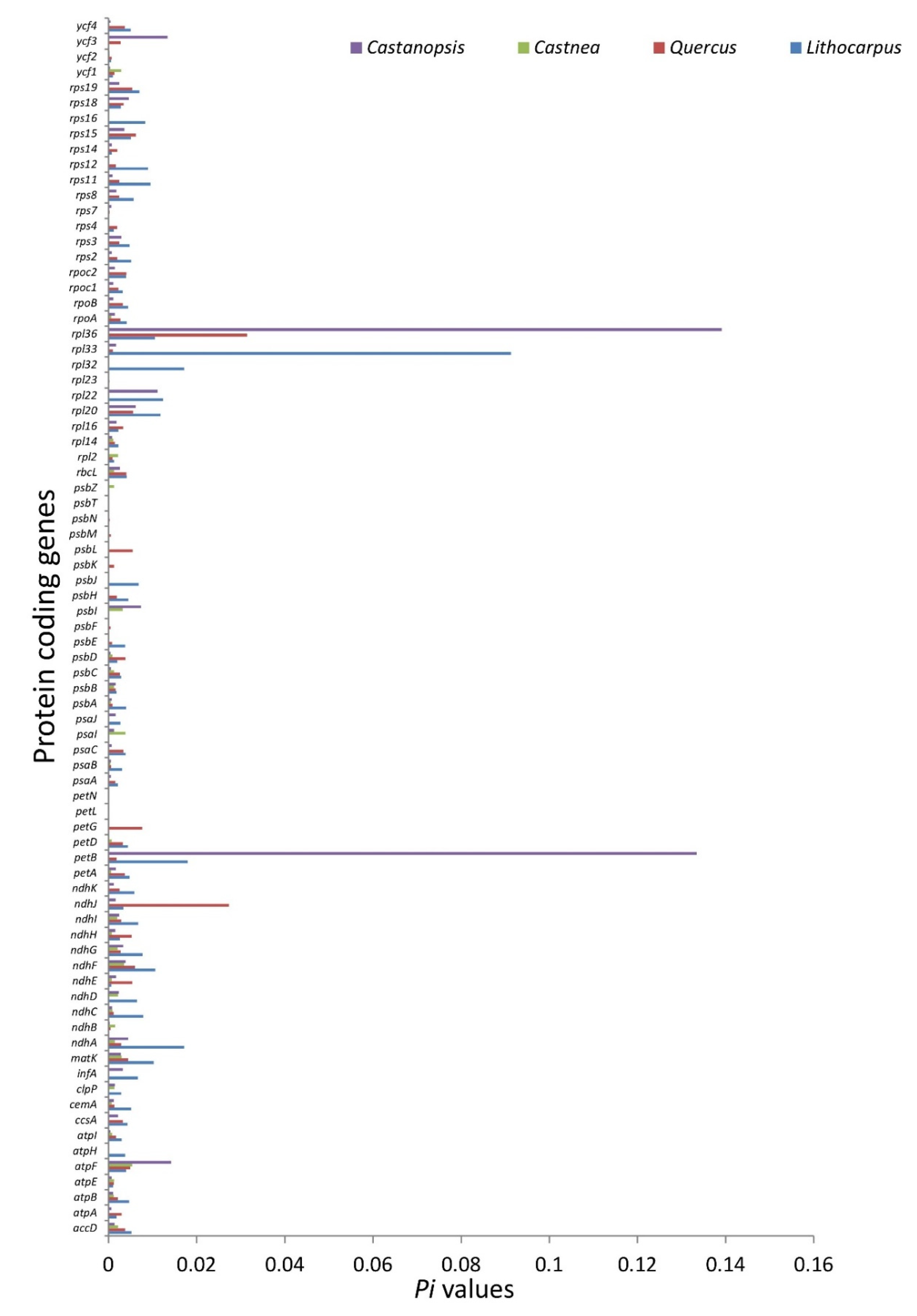
3.2.3. Nucleotide Diversity and Mutational Hotspots
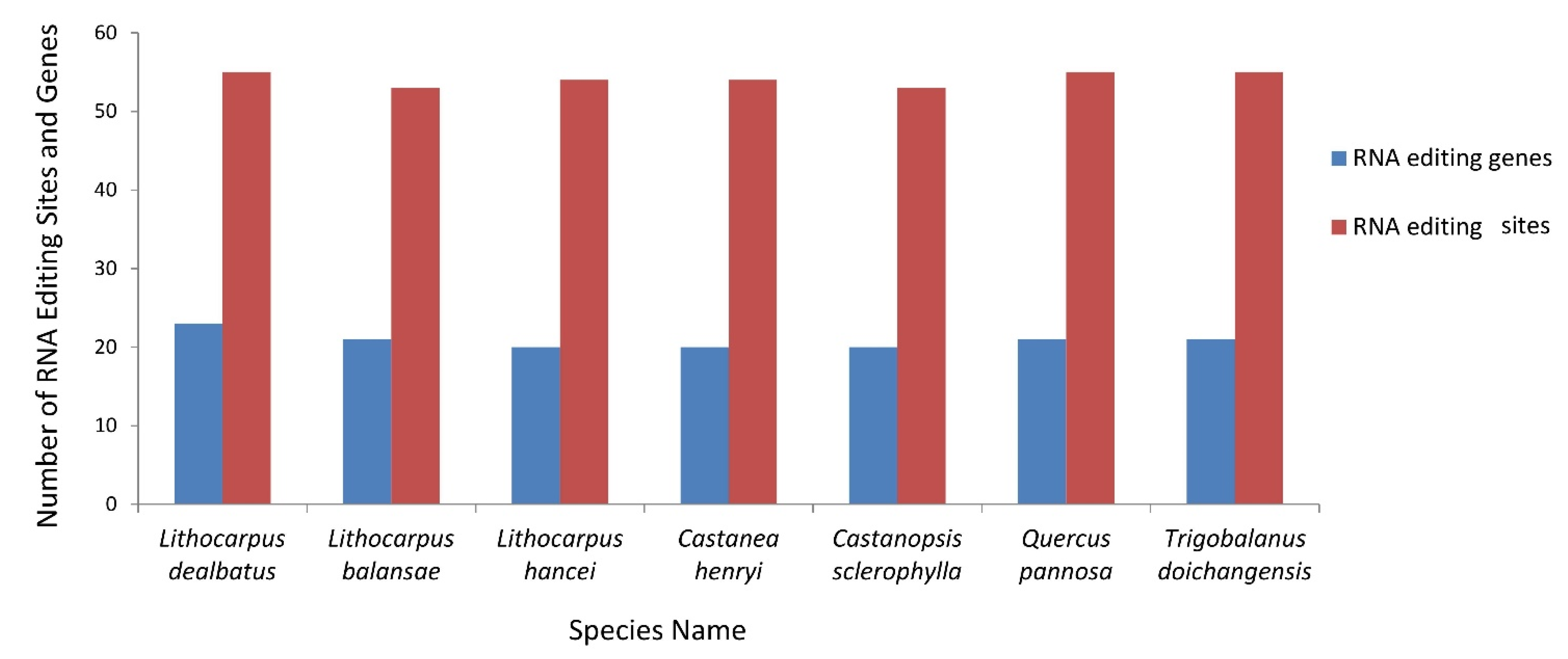
3.2.4. RNA Editing Sites and Its Comparative Analysis in Quercoideae
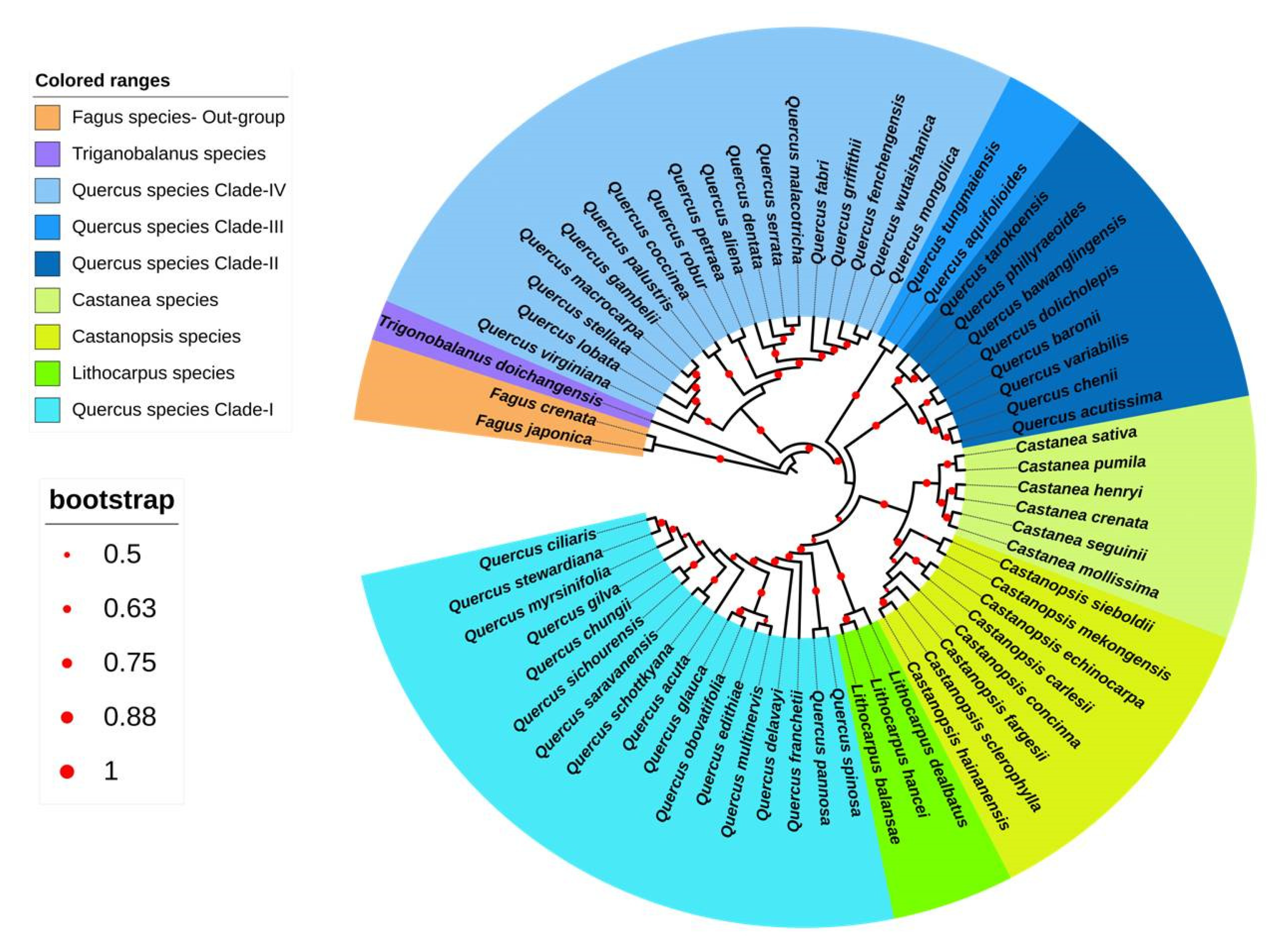
3.3. Chloroplast-Based Phylogenetic Analysis in Quercoideae
3.3.1. Phylogenetic Analysis of Quercoideae
3.3.2. Estimated Lineage (Stem) Divergence Time for Quercoideae Members
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, B.; Adhikari, D.; Singh, B.; Barik, S.K. Diversity and distribution of Fagaceae in northeastern India. In Plant and Ecosystem Diversity; Barik, S.K., Ed.; Daya Publishing House: A Division of Astral International Publication: New Delhi, India, 2022; pp. 171–178. [Google Scholar]
- Bhatt, D.; Sekar, K.C.; Kumar, K. Tree Diversity of Indian Himalayan Region; Pant National Institute of Himalayan Environment, Almora: Uttarakhand, India, 2020; ISBN 978-93-5408-274-0. [Google Scholar]
- Tang, C.Q.; Chiou, C.R.; Lin, C.T.; Lin, J.R.; Hsieh, C.F.; Tang, J.W.; Su, W.H.; Hou, X. Plant Diversity Patterns in Subtropical Evergreen Broad-Leaved Forests of Yunnan and Taiwan. Ecol. Res. 2013, 28, 81–92. [Google Scholar] [CrossRef]
- Lehmann, L.; Greijmans, M. Forests and Trees of the Central Highlands of Xieng Khouang Lao PDR; DANIDA: Vientiane, Laos, 2003; 246p. [Google Scholar]
- Singh, P.P.; Chakraborty, T.; Dermann, A.; Dermann, F.; Adhikari, D.; Gurung, P.B.; Barik, S.K.; Bauhus, J.; Fassnacht, F.E.; Dey, D.C.; et al. Assessing Restoration Potential of Fragmented and Degraded Fagaceae Forests in Meghalaya, North-East India. Forests 2020, 11, 1008. [Google Scholar] [CrossRef]
- Zhou, B.F.; Yuan, S.; Crowl, A.A.; Liang, Y.Y.; Shi, Y.; Chen, X.Y.; An, Q.Q.; Kang, M.; Manos, P.S.; Wang, B. Phylogenomic Analyses Highlight Innovation and Introgression in the Continental Radiations of Fagaceae across the Northern Hemisphere. Nat. Commun. 2022, 13, 1320. [Google Scholar] [CrossRef]
- Rao, P.; Barik, S.K.; Pandey, H.N.; Tripathi, R.S. Community Composition and Tree Population Structure in a Sub-Tropical Broad-Leaved Forest along a Disturbance Gradient. Vegetatio 1990, 88, 151–162. [Google Scholar] [CrossRef]
- Barik, S.K.; Pandey, H.N.; Tripathi, R.S.; Rao, P. Microenvironmental Variability and Species Diversity in Treefall Gaps in a Sub-Tropical Broadleaved Forest. Vegetatio 1992, 103, 31–40. [Google Scholar] [CrossRef]
- Vander Wall, S.B. The Evolutionary Ecology of Nut Dispersal. Bot. Rev. 2001, 67, 74–117. [Google Scholar] [CrossRef]
- Barik, S.K.; Tripathi, R.S.; Pandey, H.N.; Rao, P. Tree Regeneration in a Subtropical Humid Forest: Effect of Cultural Disturbance on Seed Production, Dispersal and Germination. J. Appl. Ecol. 1996, 33, 1551–1560. [Google Scholar] [CrossRef]
- Barik, S.K.; Rao, P.; Tripathi, R.S.; Pandey, H.N. Dynamics of Tree Seedling Populations in a Humid Subtropical Forest of Northeast India as Related to Disturbance. Can. J. For. Res. 1996, 26, 584–589. [Google Scholar] [CrossRef]
- Rao, P.; Barik, S.K.; Pandey, H.N.; Tripathi, R.S. Tree Seed Germination and Seedling Establishment in Treefall Gaps and Understorey in a Subtropical Forest of Northeast India. Aust. J. Ecol. 1997, 22, 136–145. [Google Scholar] [CrossRef]
- Cavender-Bares, J.; Ackerly, D.D.; Baum, D.A.; Bazzaz, F.A. Phylogenetic Overdispersion in Floridian Oak Communities. Am. Nat. 2004, 163, 823–843. [Google Scholar] [CrossRef]
- Manos, P.S.; Stanford, A.M. The Historical Biogeography of Fagaceae: Tracking the Tertiary History of Temperate and Subtropical Forests of the Northern Hemisphere. Int. J. Plant Sci. 2001, 162, S77–S93. [Google Scholar] [CrossRef]
- Moran, E.V.; Willis, J.; Clark, J.S. Genetic Evidence for Hybridization in Red Oaks (Quercus Sect. Lobatae, Fagaceae). Am. J. Bot. 2012, 99, 92–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Hu, D.; Guo, Y.; Id, R.S. Complete Chloroplast Genome of Castanopsis Sclerophylla (Lindl.) Schott: Genome Structure and Comparative and Phylogenetic Analysis. PLoS ONE 2019, 14, e0208552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, X.; Liu, H.; Wu, S.; Yuan, Y.; Li, H.; Dong, J.; Liu, Z.; An, C.; Su, Z.; Li, B. Species Identification of Oaks (Quercus L., Fagaceae) from Gene to Genome. Int. J. Mol. Sci. 2019, 20, 5940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Vázquez, L.; Chen, X.; Li, H.; Zhang, H.; Liu, Z.; Zhao, G. Development of Chloroplast and Nuclear DNA Markers for Chinese Oaks (Quercus Subgenus Quercus) and Assessment of Their Utility as DNA Barcodes. Front. Plant Sci. 2017, 8, 816. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Qu, X.J.; Zhang, R.; Stull, G.W.; Yi, T.S. Plastid Phylogenomic Analyses of Fagales Reveal Signatures of Conflict and Ancient Chloroplast Capture. Mol. Phylogenet. Evol. 2021, 163, 107232. [Google Scholar] [CrossRef]
- Piredda, R.; Simeone, M.C.; Attimonelli, M.; Bellarosa, R.; Schirone, B. Prospects of Barcoding the Italian Wild Dendroflora: Oaks Reveal Severe Limitations to Tracking Species Identity. Mol. Ecol. Resour. 2011, 11, 72–83. [Google Scholar] [CrossRef]
- Zhou, H.; Gao, X.; Woeste, K.; Zhao, P.; Zhang, S. Comparative Analysis of the Complete Chloroplast Genomes of Four Chestnut Species (Castanea). Forests 2021, 12, 861. [Google Scholar] [CrossRef]
- Song, Y.; Wang, S.; Ding, Y.; Xu, J.; Li, M.F.; Zhu, S.; Chen, N. Chloroplast Genomic Resource of Paris for Species Discrimination. Sci. Rep. 2017, 7, 3427. [Google Scholar] [CrossRef] [Green Version]
- Cosner, M.E.; Raubeson, L.A.; Jansen, R.K. Chloroplast DNA Rearrangements in Campanulaceae: Phylogenetic Utility of Highly Rearranged Genomes. BMC Evol. Biol. 2004, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast Genomes: Diversity, Evolution, and Applications in Genetic Engineering. Genome Biol. 2016, 17, 1–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nock, C.J.; Waters, D.L.E.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast Genome Sequences from Total DNA for Plant Identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Rogalski, M.; Vieira, L.D.N.; Fraga, H.P.; Guerra, M.P. Plastid Genomics in Horticultural Species: Importance and Applications for Plant Population Genetics, Evolution, and Biotechnology. Front. Plant Sci. 2015, 6, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; dePamphilis, C.W.; Müller, K.F.; Quandt, D. The Evolution of the Plastid Chromosome in Land Plants: Gene Content, Gene Order, Gene Function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- de Santana Lopes, A.; Pacheco, T.G.; dos Santos, K.G.; do Vieira, L.N.; Guerra, M.P.; Nodari, R.O.; de Souza, E.M.; de Oliveira Pedrosa, F.; Rogalski, M. The Linum Usitatissimum L. Plastome Reveals Atypical Structural Evolution, New Editing Sites, and the Phylogenetic Position of Linaceae within Malpighiales. Plant Cell Rep. 2018, 37, 307–328. [Google Scholar] [CrossRef]
- Mo, Z.; Lou, W.; Chen, Y.; Jia, X.; Zhai, M.; Guo, Z.; Xuan, J. The Chloroplast Genome of Carya Illinoinensis: Genome Structure, Adaptive Evolution, and Phylogenetic Analysis. Forests 2020, 11, 207. [Google Scholar] [CrossRef] [Green Version]
- Menezes, A.P.A.; Resende-Moreira, L.C.; Buzatti, R.S.O.; Nazareno, A.G.; Carlsen, M.; Lobo, F.P.; Kalapothakis, E.; Lovato, M.B. Chloroplast Genomes of Byrsonima Species (Malpighiaceae): Comparative Analysis and Screening of High Divergence Sequences. Sci. Rep. 2018, 8, 2210. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chang, E.; Liu, J.; Huang, Y.; Wang, Y.; Yao, N.; Jiang, Z.-P. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus Bawanglingensis Huang, Li et Xing, a Vulnerable Oak Tree in China. Forests 2019, 10, 587. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Asaf, S.; Khan, A.L.; Shehzad, T.; Al-Rawahi, A.; Al-Harrasi, A. Comparative Chloroplast Genomics of Endangered Euphorbia Species: Insights into Hotspot Divergence, Repetitive Sequence Variation, and Phylogeny. Plants 2020, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Sun, Y.; Liu, J.; Xu, C.; Zou, X.; Chen, X.; Liu, Y.; Wu, P.; Yang, X.; Zhou, S. DNA Barcoding of Oryza: Conventional, Specific, and Super Barcodes. Plant Mol. Biol. 2021, 105, 215–228. [Google Scholar] [CrossRef]
- Ma, C.; Yan, H.; Ge, X.; Zhejiang, F. The Complete Chloroplast Genome of Lithocarpus Hancei ( Benth.) Rehd ( Fagaceae ) from Zhejiang, China. Mitochondrial DNA Part B 2022, 6, 2022–2023. [Google Scholar] [CrossRef] [PubMed]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De Novo Assembly of Organelle Genomes from Whole Genome Data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and Accurate Annotation of Organelle Genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Bi, G.; Mao, Y.; Xing, Q.; Cao, M. HomBlocks: A Multiple-Alignment Construction Pipeline for Organelle Phylogenomics Based on Locally Collinear Block Searching. Genomics 2018, 110, 18–22. [Google Scholar] [CrossRef]
- Suchiang, B.R.; Nonghuloo, I.M.; Kharbhih, S.; Singh, P.P.; Tiwary, R.; Adhikari, D.; Upadhaya, K.; Ramanujam, P.; Barik, S.K. Tree Diversity and Community Composition in Sacred Forests Are Superior than the Other Community Forests in a Human-Dominated Landscape of Meghalaya. Trop. Ecol. 2020, 61, 84–105. [Google Scholar] [CrossRef]
- Mohanta, T.K.; Mishra, A.K.; Khan, A.; Hashem, A.; Abd_Allah, E.F.; Al-Harrasi, A. Gene Loss and Evolution of the Plastome. Genes 2020, 11, 1133. [Google Scholar] [CrossRef]
- Frailey, D.C.; Chaluvadi, S.R.; Vaughn, J.N.; Coatney, C.G.; Bennetzen, J.L. Gene Loss and Genome Rearrangement in the Plastids of Five Hemiparasites in the Family Orobanchaceae. BMC Plant Biol. 2018, 18, 30. [Google Scholar] [CrossRef] [Green Version]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many Parallel Losses of InfA from Chloroplast DNA during Angiosperm Evolution with Multiple Independent. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Asaf, S.; Khan, A.L.; Khan, A.; Khan, G.; Lee, I. Expanded Inverted Repeat Region with Large Scale Inversion in the First Complete Plastid Genome Sequence of Plantago Ovata. Sci. Rep. 2020, 10, 3881. [Google Scholar] [CrossRef] [Green Version]
- He, P.; Huang, S.; Xiao, G.; Zhang, Y.; Yu, J. Abundant RNA Editing Sites of Chloroplast Protein-Coding Genes in Ginkgo Biloba and an Evolutionary Pattern Analysis. BMC Plant Biol. 2016, 16, 257. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.-M.; Jin, J.; Yi, T. Plastome Structural Conservation and Evolution in the Clusioid Clade of Malpighiales. Sci. Rep. 2020, 10, 9091. [Google Scholar] [CrossRef] [PubMed]
- Abdullah; Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast Genome of Hibiscus Rosa-Sinensis (Malvaceae): Comparative Analyses and Identification of Mutational Hotspots. Genomics 2020, 112, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, Y.; Xu, J.; Li, W.; Li, M.F. Characterization of the Complete Chloroplast Genome Sequence of Dalbergia Species and Its Phylogenetic Implications. Sci. Rep. 2019, 9, 20401. [Google Scholar] [CrossRef] [PubMed]
- Nie, X.; Lv, S.; Zhang, Y.; Du, X.; Wang, L.; Biradar, S.S.; Tan, X.; Wan, F.; Weining, S. Complete Chloroplast Genome Sequence of a Major Invasive Species, Crofton Weed (Ageratina Adenophora). PLoS ONE 2012, 7, e36869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.; Civitello, A.; Hammond, H.A.; Caskey, C.T. DNA Typing and Genetic Mapping with Trimeric and Tetrameric Tandem Repeats. Am. J. Hum. Genet. 1991, 49, 746–756. [Google Scholar]
- Li, B.; Zheng, Y. Dynamic Evolution and Phylogenomic Analysis of the Chloroplast Genome in Schisandraceae. Sci. Rep. 2018, 8, 9285. [Google Scholar] [CrossRef]
- Shelke, R.G.; Basak, S.; Rangan, L. Development of EST-SSR Markers for Pongamia Pinnata by Transcriptome Database Mining: Cross-Species Amplification and Genetic Diversity. Physiol. Mol. Biol. Plants 2020, 26, 2225–2241. [Google Scholar] [CrossRef]
- Rajendrakumar, P.; Biswal, A.K.; Balachandran, S.M.; Srinivasarao, K.; Sundaram, R.M. Simple Sequence Repeats in Organellar Genomes of Rice: Frequency and Distribution in Genic and Intergenic Regions. Bioinformatics 2007, 23, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Tambarussi, E.V.; Melotto-Passarin, D.M.; Gonzalez, S.G.; Brigati, J.B.; Jesus, F.A.; Barbosa, A.L.; Dressano, K.; Carrer, H. In Silico Analysis of Simple Sequence Repeats from Chloroplast Genomes of Solanaceae Species. Crop Breed. Appl. Biotechnol. 2009, 9, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [Green Version]
- Freyer, R.; Kiefer-Meyer, M.-C.; Kossel, H. Occurrence of Plastid RNA Editing in All Major Lineages of Land Plants. Proc. Natl. Acad. Sci. USA 1997, 94, 6285–6290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, N.F.; Christoff, A.P.; da Fonseca, G.C.; Kulcheski, F.R.; Margis, R. Unveiling Chloroplast RNA Editing Events Using Next Generation Small RNA Sequencing Data. Front. Plant Sci. 2017, 8, 1686. [Google Scholar] [CrossRef] [Green Version]
- Small, I.D.; Schallenberg-Rüdinger, M.; Takenaka, M.; Mireau, H.; Ostersetzer-Biran, O. Plant Organellar RNA Editing: What 30 Years of Research Has Revealed. Plant J. 2020, 101, 1040–1056. [Google Scholar] [CrossRef] [PubMed]
- Introduction. In The Neutral Theory of Molecular Evolution; Kimura, M. (Ed.) Cambridge University Press: Cambridge, UK, 1983; pp. xi–xvi. ISBN 9780521317931. [Google Scholar]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit Incorporating Gamma-Series Methods and Sliding Window Strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hu, Y.; Ren, T.; Sun, J.; Zhao, G. Remarkably Conserved Plastid Genomes of Quercus Group Cerris in China: Comparative and Phylogenetic Analyses. Nord. J. Bot. 2018, 36, e01921. [Google Scholar] [CrossRef]
- Xu, X.; Wang, D. Comparative Chloroplast Genomics of Corydalis Species (Papaveraceae): Evolutionary Perspectives on Their Unusual Large Scale Rearrangements. Front. Plant Sci. 2021, 11, 600354. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Chakrabartty, I.; Hedaoo, V.; Shelke, R.G.; Rangan, L. Assessment of genetic variation among wild Alpinia nigra (Zingiberaceae) population: An approach based on molecular phylogeny. Mol. Biol. Rep. 2019, 46, 177–189. [Google Scholar] [CrossRef]
- Xu, J.; Shen, X.; Liao, B.; Xu, J.; Hou, D. Comparing and Phylogenetic Analysis Chloroplast Genome of Three Achyranthes Species. Sci. Rep. 2020, 10, 10818. [Google Scholar] [CrossRef]
- Larson-Johnson, K. Phylogenetic Investigation of the Complex Evolutionary History of Dispersal Mode and Diversification Rates across Living and Fossil Fagales. New Phytol. 2016, 209, 418–435. [Google Scholar] [CrossRef]
- Manos, P.S.; Cannon, C.H.; Oh, S.-H. Phylogenetic Relationships and Taxonomic Status Of the Paleoendemic Fagaceae Of Western North America: Recognition Of A New Genus, Notholithocarpus. Madroño 2008, 55, 181–190. [Google Scholar] [CrossRef]
- Zhe-Kun, Z. Fossils of the Fagaceae and their implications in systematics and biogeography. J. Syst. Evol. 1999, 37, 369. [Google Scholar]
- Yang, J.; Huang, Y.; Yang, Z.; Zhou, C.; Hu, X. Identification and Quantitative Evaluation of Major Sweet Ingredients in Sweet Tea (Lithocarpus Polystachyus Rehd.) Based Upon Location, Harvesting Time, Leaf Age. J. Chem. Soc. Pak. 2018, 40, 158–164. [Google Scholar]
- Whittemore, A.T.; Schaal, B.A. Interspecific Gene Flow in Sympatric Oaks. Proc. Natl. Acad. Sci. USA 1991, 88, 2540–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieseberg, L.H.; Soltis, D.E. Phylogenetic Consequences of Cytoplasmic. Evol. Trends Plants 1991, 5, 65–84. [Google Scholar]
- Oh, S.H.; Manos, P.S. Molecular Phylogenetics and Cupule Evolution in Fagaceae as Inferred from Nuclear CRABS CLAW Sequences. Taxon 2008, 57, 434–451. [Google Scholar]
- Denk, T.; Grimm, G.W. The Oaks of Western Eurasia: Traditional Classifications and Evidence from Two Nuclear Markers. Taxon 2010, 59, 351–366. [Google Scholar] [CrossRef]
- Hubert, F.; Grimm, G.W.; Jousselin, E.; Berry, V.; Franc, A.; Kremer, A. Multiple Nuclear Genes Stabilize the Phylogenetic Backbone of the Genus Quercus. Syst. Biodivers. 2014, 12, 405–423. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, W.; Li, R.-Q.; Lin, L.; Liu, Y.; Zhou, Z.-K.; Li, Z.Y.; Chen, Z.D. Large-scale phylogenetic analyses reveal fagalean diversification promoted by the interplay of diaspores and environments in the Paleogene. Perspect Plant Ecol. Evol. Syst. 2014, 16, 101–110. [Google Scholar] [CrossRef]
- Aldrich, P.R.; Cavender-bares, J. Wild Crop Relatives: Genomic and Breeding Resources; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar] [CrossRef]
- Stegemann, S.; Keuthe, M.; Greiner, S.; Bock, R. Horizontal Transfer of Chloroplast Genomes between Plant Species. Proc. Natl. Acad. Sci. USA 2012, 109, 2434–2438. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.B.; Campbell, C.S.; Hong, D.Y.; Wen, J. Phylogenetic Relationships and Chloroplast Capture in the Amelanchier-Malacomeles-Peraphyllum Clade (Maleae, Rosaceae): Evidence from Chloroplast Genome and Nuclear Ribosomal DNA Data Using Genome Skimming. Mol. Phylogenet. Evol. 2020, 147, 106784. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Category | Gene Name | ||||
|---|---|---|---|---|---|
| Ribosomal RNA genes | rrn16 D | rrn23 D | rrn4.5 D | rrn5 D | |
| Transfer RNA genes | trnA-UGC D * | trnC-GCA | trnD-GUC | trnE-UUC | trnF-GAA |
| trnfM-CAU | trnG-GCC D * | trnH-GUG | trnI-CAU D | trnI-GAU D * | |
| trnK-UUU * | trnL-CAA D | trnL-UAA * | trnL-UAG | trnM-CAU | |
| trnN-GUU D | trnP-GGG | trnP-UGG | trnQ-UUG | trnR-ACG D | |
| trnR-UCU | trnS-GCU | trnS-GGA | trnS-UGA | trnT-GGU D | |
| trnT-UGU | trnV-GAC D | trnV-UAC * | trnW-CCA | trnY-GUA | |
| Large subunit of ribosomal proteins (LSU) | rpl2 D * | rpl14 | rpl16 | rpl20 | rpl22 |
| rpl23 D | rpl32 | rpl33 | rpl36 | ||
| Small subunit of ribosomal proteins (SSU) | rps2 | rps3 | rps4 | rps7 D | rps8 |
| rps11 | rps12 D * | rps14 | rps15 | rps16 * | |
| rps18 | rps19 | ||||
| DNA dependant RNA polymerase | rpoA | rpoB | rpoC1 * | rpoC2 | |
| Photosystem I | psaA | psaB | psaC | psaI | psaJ |
| Photosystem II | psbA | psbB | psbC | psbDD | psbE |
| psbF | psbH | psbI | psbJ | psbK | |
| psbL | psbM | psbN | psbT | psbZ | |
| Cytochrome b/f | petA | petB * | petD | petG | petL |
| petN | |||||
| ATP synthase | atpA | atpB | atpE | atpF * | atpH |
| atpI | |||||
| Protease | clpP ** | ||||
| Rubisco | rbcL | ||||
| NADH dehydrogenase | ndhA * | ndhB D * | ndhC | ndhD | ndhE |
| ndhF | ndhG | ndhH | ndhI | ndhJ | |
| ndhK | |||||
| Maturase | matK | ||||
| Envelop membrane protein | cemA | ||||
| Subunit of acetyl-CoA-carboxylase | accD | ||||
| C-type cytochrome synthesis gene | ccsA | ||||
| Conserved hypothetical chloroplast open reading frames | ycf1 D | ycf2 D | ycf3 ** | ycf4 | |
| Species | L. dealbatus | L. balansae | L. hancei | C. henryi | C. sclerophylla | Q. pannosa | T. doichangensis |
|---|---|---|---|---|---|---|---|
| Genome size | 161,476 bp | 161,020 bp | 161,304 bp | 160,907 bp | 160,497 bp | 161,222 bp | 159,938 bp |
| LSC | 90,732 bp | 90,596 bp | 90,585 bp | 90,527 bp | 90, 255 bp | 90,522 bp | 89,445 bp |
| SSC | 18,987 bp | 19,160 bp | 18,959 bp | 18,998 bp | 25,675 bp | 19,000 bp | 19,295 bp |
| IR | 25,879 bp | 25,632 bp | 25,880 bp | 25,961 bp | 18,892 bp | 25,850 bp | 25,600 bp |
| Protein coding genes | 86 | 87 | 87 | 82 | 86 | 85 | 81 |
| rRNAs | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| tRNAs | 39 | 39 | 44 | 37 | 37 | 37 | 39 |
| Duplicated genes | 21 | 19 | 24 | 17 | 21 | 17 | 17 |
| CDS | 80,577 bp | 80,142 bp | 80,199 bp | 77,685 bp | 79,647 bp | 78,852 bp | 71,778 bp |
| NCDS | 80,899 bp | 80,878 bp | 81,105 bp | 83,222 bp | 80,850 bp | 82,370 bp | 88,360 bp |
| GC% | 36.7 | 36.7 | 36.7 | 36.7 | 36.8 | 36.9 | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shelke, R.G.; Banerjee, R.P.; Joshi, B.; Singh, P.P.; Tiwari, G.J.; Adhikari, D.; Jena, S.N.; Barik, S.K. Chloroplast Genome of Lithocarpus dealbatus (Hook.f. & Thomson ex Miq.) Rehder Establishes Monophyletic Origin of the Species and Reveals Mutational Hotspots with Taxon Delimitation Potential. Life 2022, 12, 828. https://doi.org/10.3390/life12060828
Shelke RG, Banerjee RP, Joshi B, Singh PP, Tiwari GJ, Adhikari D, Jena SN, Barik SK. Chloroplast Genome of Lithocarpus dealbatus (Hook.f. & Thomson ex Miq.) Rehder Establishes Monophyletic Origin of the Species and Reveals Mutational Hotspots with Taxon Delimitation Potential. Life. 2022; 12(6):828. https://doi.org/10.3390/life12060828
Chicago/Turabian StyleShelke, Rahul Gunvantrao, Rudra Prasad Banerjee, Babita Joshi, Prem Prakash Singh, Gopal Ji Tiwari, Dibyendu Adhikari, Satya Narayan Jena, and Saroj Kanta Barik. 2022. "Chloroplast Genome of Lithocarpus dealbatus (Hook.f. & Thomson ex Miq.) Rehder Establishes Monophyletic Origin of the Species and Reveals Mutational Hotspots with Taxon Delimitation Potential" Life 12, no. 6: 828. https://doi.org/10.3390/life12060828