Distinct Microbial Community of Phyllosphere Associated with Five Tropical Plants on Yongxing Island, South China Sea

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
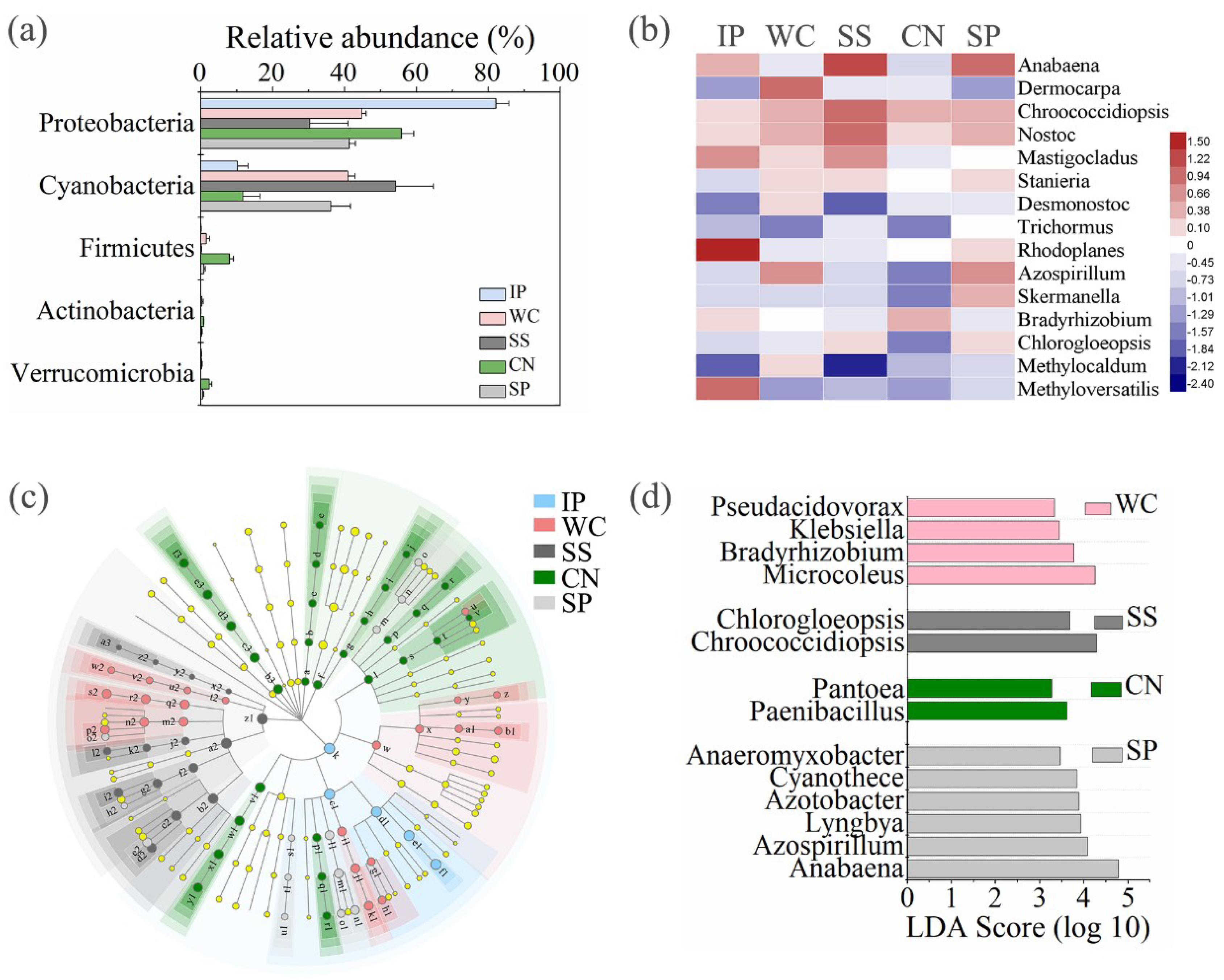
3.1. Quantitative Abundance
3.2. Alpha Diversity
3.3. Beta Diversity
3.4. Sequence Variants and Dominant Genera
3.4.1. Fungal Sequences
3.4.2. Bacterial Sequences
3.4.3. Diazotrophic Sequences
3.5. Predictive Metagenome Analysis
4. Discussion
4.1. Quantitative Abundance and Diversity of Microbial Communities among Five Plant Species
4.2. Fungal Communities among Five Plant Species
4.3. Bacterial Communities among Five Plant Species
4.4. Diazotrophic Communities among Five Plant Species
4.5. Predictive Metabolism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lindow, S.E.; Leveau, J.H. Phyllosphere microbiology. Curr. Opin. Biotechnol. 2002, 13, 238–243. [Google Scholar] [CrossRef]
- Beattie, G.A.; Lindow, S.E. Bacterial colonization of leaves: A spectrum of strategies. Phytopathology 1999, 89, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Kishore, G.K.; Pande, S.; Podile, A.R. Biological control of late leaf spot of peanut (arachis hypogaea) with chitinolytic bacteria. Phytopathology 2005, 95, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Lindow, S.E.; Brandl, M.T. Microbiology of the phyllosphere. Appl. Environ. Microbiol. 2003, 69, 1875–1883. [Google Scholar] [CrossRef]
- Terhonen, E.; Blumenstein, K.; Kovalchuk, A.; Asiegbu, F.O. Forest tree microbiomes and associated fungal endophytes: Functional roles and impact on forest health. Forests 2019, 10, 32. [Google Scholar] [CrossRef]
- Porras-Alfaro, A.; Bayman, P. Hidden fungi, emergent properties: Endophytes and microbiomes. In Annual Review of Phytopathology; VanAlfen, N.K., Bruening, G., Leach, J.E., Eds.; Annual Reviews: Palo Alto, CA, USA, 2011; Volume 49, pp. 291–315. [Google Scholar]
- Innerebner, G.; Knief, C.; Vorholt, J.A. Protection of arabidopsis thaliana against leaf-pathogenic pseudomonas syringae by sphingomonas strains in a controlled model system. Appl. Environ. Microbiol. 2011, 77, 3202–3210. [Google Scholar] [CrossRef]
- Lugtenberg, B.J.J.; de Weger, L.A.; Bennett, J.W. Microbial stimulation of plant growth and protection from disease. Curr. Opin. Biotechnol. 1991, 2, 457–464. [Google Scholar] [CrossRef]
- Kinkel, L.L.; Wilson, M.; Lindow, S.E. Plant species and plant incubation conditions influence variability in epiphytic bacterial population size. Microb. Ecol. 2000, 39, 1–11. [Google Scholar] [CrossRef]
- Hardoim, P.R.; van Overbeek, L.S.; Berg, G.; Pirttila, A.M.; Compant, S.; Campisano, A.; Doring, M.; Sessitsch, A. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef]
- Dulla, G.; Marco, M.; Quinones, B.; Lindow, S. A closer look at pseudomonas syringae as a leaf colonist- the pathogen p-syringae thrives on healthy plants by employing quorum sensing, virulence factors, and other traits. Asm News 2005, 71, 469. [Google Scholar]
- Kovalchuk, A.; Mukrimin, M.; Zeng, Z.; Raffaello, T.; Liu, M.X.; Kasanen, R.; Sun, H.; Asiegbu, F.O. Mycobiome analysis of asymptomatic and symptomatic norway spruce trees naturally infected by the conifer pathogens Heterobasidion spp. Environ. Microbiol. Rep. 2018, 10, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Papen, H.; Geβler, A.; Zumbusch, E.; Rennenberg, H. Chemolithoautotrophic nitrifiers in the phyllosphere of a spruce ecosystem receiving high atmospheric nitrogen input. Curr. Microbiol. 2002, 44, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Fuernkranz, M.; Wanek, W.; Richter, A.; Abell, G.; Rasche, F.; Sessitsch, A. Nitrogen fixation by phyllosphere bacteria associated with higher plants and their colonizing epiphytes of a tropical lowland rainforest of costa rica. ISME J. 2008, 2, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Van Aken, B.; Peres, C.M.; Doty, S.L.; Yoon, J.M.; Schnoor, J.L. Methylobacterium populi sp nov., a novel aerobic, pink-pigmented, facultatively methylotrophic, methane-utilizing bacterium isolated from poplar trees (populus deltoides x nigra dn34). Int. J. Syst. Evol. Microbiol. 2004, 54, 1191–1196. [Google Scholar] [CrossRef]
- Baldrian, P. Forest microbiome: Diversity, complexity and dynamics. Fems Microbiol. Rev. 2016, 41, 109–130. [Google Scholar] [CrossRef]
- Redford, A.J.; Bowers, R.M.; Knight, R.; Linhart, Y.; Fierer, N. The ecology of the phyllosphere: Geographic and phylogenetic variability in the distribution of bacteria on tree leaves. Environ. Microbiol. 2010, 12, 2885–2893. [Google Scholar] [CrossRef]
- Kembel, S.W.; O’Connor, T.K.; Arnold, H.K.; Hubbell, S.P.; Wright, S.J.; Green, J.L. Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest. Proc. Natl. Acad. Sci. USA 2014, 111, 13715–13720. [Google Scholar] [CrossRef] [Green Version]
- Jumpponen, A.; Jones, K.L. Massively parallel 454 sequencing indicates hyperdiverse fungal communities in temperate quercus macrocarpa phyllosphere. New Phytol. 2009, 184, 438–448. [Google Scholar] [CrossRef]
- Abril, A.B.; Torres, P.A.; Bucher, E.H. The importance of phyllosphere microbial populations in nitrogen cycling in the chaco semi-arid woodland. J. Trop. Ecol. 2005, 21, 103–107. [Google Scholar] [CrossRef]
- Cleveland, C.C.; Townsend, A.R.; Schimel, D.S.; Fisher, H.; Howarth, R.W.; Hedin, L.O.; Perakis, S.S.; Latty, E.F.; Fischer, J.C.V.; Elseroad, A.; et al. Global patterns of terrestrial biological nitrogen (n2) fixation in natural ecosystems. Glob. Biogeochem. Cycles 1999, 13, 623–645. [Google Scholar] [CrossRef]
- Roggy, J.C.; Prevost, M.F.; Garbaye, J.; Domenach, A.M. Nitrogen cycling in the tropical rain forest of french guiana: Comparison of two sites with contrasting soil types using delta n-15. J. Trop. Ecol. 1999, 15, 1–22. [Google Scholar] [CrossRef]
- Poly, F.; Monrozier, L.J.; Bally, R. Improvement in the rflp procedure for studying the diversity of nifh genes in communities of nitrogen fixers in soil. Res. Microbiol. 2001, 152, 95–103. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Asshauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4fun: Predicting functional profiles from metagenomic 16s rrna data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.C.; Dobson, A.; O’Sullivan, O.; Crispie, F.; Fouhy, F.; Cotter, P.D.; Shanahan, F.; Kiely, B.; Hill, C.; Ross, R.P. Effect of broad- and narrow-spectrum antimicrobials on clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. USA 2011, 108, 4639–4644. [Google Scholar] [CrossRef] [PubMed]
- Heip, C. A new index measuring evenness. J. Mar. Biol. Assoc. UK 1974, 54, 555–557. [Google Scholar] [CrossRef]
- Gotelli, N.J.; Colwell, R.K. Estimating species richness. Biol. Divers. Front. Meas. Assess. 2011, 12, 39–54. [Google Scholar]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.P.; Clement, C.; Ouhdouch, Y.; van Wezel, G.P. Cd taxonomy, physiology, and natural products of actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–43. [Google Scholar] [CrossRef]
- Ellison, J.C. Pacific island beaches: Values, threats and rehabilitation. In Beach Management Tools-Concepts, Methodologies and Case Studies; Botero, C.M., Cervantes, O., Finkl, C.W., Eds.; Springer International Publishing: Berlin, Germany, 2018; pp. 679–700. [Google Scholar]
- Lindow, S.E.; Andersen, G.L. Influence of immigration on epiphytic bacterial populations on navel orange leaves. Appl. Environ. Microbiol. 1996, 62, 2978–2987. [Google Scholar] [Green Version]
- Yadav, R.K.P.; Karamanoli, K.; Vokou, D. Bacterial colonization of the phyllosphere of mediterranean perennial species as influenced by leaf structural and chemical features. Microb. Ecol. 2005, 50, 185–196. [Google Scholar] [CrossRef]
- Bloom, A.J.; Chapin, F.S.; Mooney, H.A. Resource limitation in plants-an economic analogy. Annu. Rev. Ecol. Syst. 1985, 16, 363–392. [Google Scholar] [CrossRef]
- Ruppel, S.; Krumbein, A.; Schreiner, M. Composition of the phyllospheric microbial populations on vegetable plants with different glucosinolate and carotenoid compositions. Microb. Ecol. 2008, 56, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Sun, X.; He, C.; Maitra, P.; Li, X.-C.; Guo, L.-D. Phyllosphere epiphytic and endophytic fungal community and network structures differ in a tropical mangrove ecosystem. Microbiome 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.J.; White, J.F.; Arnold, A.E.; Redman, R.S. Fungal endophytes: Diversity and functional roles. New Phytol. 2009, 182, 314–330. [Google Scholar] [CrossRef]
- Shah, A.; Hassan, Q.P.; Mushtaq, S.; Shah, A.M.; Hussain, A. Chemoprofile and functional diversity of fungal and bacterial endophytes and role of ecofactors—A review. J. Basic Microbiol. 2017, 57, 814–826. [Google Scholar] [CrossRef]
- Osono, T.; Mori, A. Colonization of japanese beech leaves by phyllosphere fungi. Mycoscience 2003, 44, 437–441. [Google Scholar] [CrossRef]
- Kirk, P.M.; Cannon, P.F.; Minter, D.W.; Stalpers, J.A. Dictionary of the Fungi, 10th ed.; CAB International: Wallingford, UK, 2008; p. 771. [Google Scholar]
- Tudzynski, B. Biosynthesis of gibberellins in gibberella fujikuroi: Biomolecular aspects. Appl. Microbiol. Biotechnol. 1999, 52, 298–310. [Google Scholar] [CrossRef]
- Schardl, C.L.; Young, C.A.; Hesse, U.; Amyotte, S.G.; Andreeva, K.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Moore, N.; Oeser, B.; et al. Plant-symbiotic fungi as chemical engineers: Multi-genome analysis of the clavicipitaceae reveals dynamics of alkaloid loci. Plos Genet. 2013, 9, 26. [Google Scholar] [CrossRef]
- Kadivar, H.; Stapleton, A.E. Ultraviolet radiation alters maize phyllosphere bacterial diversity. Microb. Ecol. 2003, 45, 353–361. [Google Scholar] [CrossRef]
- Tosi, S.; Onofri, S.; Brusoni, M.; Zucconi, L.; Vishniac, H. Response of antarctic soil fungal assemblages to experimental warming and reduction of uv radiation. Polar Biol. 2005, 28, 470–482. [Google Scholar] [CrossRef]
- Ruibal, C.; Gueidan, C.; Selbmann, L.; Gorbushina, A.A.; Crous, P.W.; Groenewald, J.Z.; Muggia, L.; Grube, M.; Isola, D.; Schoch, C.L.; et al. Phylogeny of rock-inhabiting fungi related to dothideomycetes. Stud. Mycol. 2009, 64, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Sundin, G.W.; Jacobs, J.L. Ultraviolet radiation (uvr) sensitivity analysis and uvr survival strategies of a bacterial community from the phyllosphere of field-grown peanut (arachis hypogeae l.). Microb. Ecol. 1999, 38, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A.; Lawley, B.; Newsham, K.K. Solar uv-b radiation inhibits the growth of antarctic terrestrial fungi. Appl. Environ. Microbiol. 2003, 69, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Kembel, S.W.; Mueller, R.C. Plant traits and taxonomy drive host associations in tropical phyllosphere fungal communities. Botany 2014, 92, 9. [Google Scholar] [CrossRef]
- Geiser, D.M.; Gueidan, C.; Miadlikowska, J.; Lutzoni, F.; Kauff, F.; Hofstetter, V.; Fraker, E.; Schoch, C.L.; Tibell, L.; Untereiner, W.A.; et al. Eurotiomycetes: Eurotiomycetidae and chaetothyriomycetidae. Mycologia 2006, 98, 1053–1064. [Google Scholar] [CrossRef]
- Ensminger, P.A. Control of development in plants and fungi by far-uv radiation. Physiol. Plant. 1993, 88, 501–508. [Google Scholar] [CrossRef]
- Kim, M.; Singh, D.; Lai-Hoe, A.; Go, R.; Abdul Rahim, R.; Ainuddin, A.N.; Chun, J.; Adams, J.M. Distinctive phyllosphere bacterial communities in tropical trees. Microb. Ecol. 2012, 63, 674–681. [Google Scholar] [CrossRef]
- Ugarelli, K.; Laas, P.; Stingl, U. The microbial communities of leaves and roots associated with turtle grass (thalassia testudinum) and manatee grass (syringodium filliforme) are distinct from seawater and sediment communities, but are similar between species and sampling sites. Microorganisms 2019, 7, 20. [Google Scholar] [CrossRef]
- De Maayer, P.; Chan, W.Y.; Blom, J.; Venter, S.N.; Duffy, B.; Smits, T.H.M.; Coutinho, T.A. The large universal pantoea plasmid lpp-1 plays a major role in biological and ecological diversification. BMC Genom. 2012, 13, 12. [Google Scholar] [CrossRef]
- Van Laere, K.M.J.; Hartemink, R.; Bosveld, M.; Schols, H.A.; Voragen, A.G.J. Fermentation of plant cell wall derived polysaccharides and their corresponding oligosaccharides by intestinal bacteria. J. Agric. Food Chem. 2000, 48, 1644–1652. [Google Scholar] [CrossRef]
- Gourion, B.; Rossignol, M.; Vorholt, J.A. A proteomic study of methylobacterium extorquens reveals a response regulator essential for epiphytic growth. Proc. Natl. Acad. Sci. USA 2006, 103, 13186–13191. [Google Scholar] [CrossRef] [PubMed]
- Liping, W.; Wanpeng, W.; Qiliang, L.; Zongze, S. Gene diversity of cyp153a and alkb alkane hydroxylases in oil-degrading bacteria isolated from the atlantic ocean. Environ. Microbiol. 2010, 12, 1230–1242. [Google Scholar]
- Shannon, M.C. The Effects of Salinity on Cellular and Biochemical Processes Associated with Salt Tolerance in Tropical Plants; Usda/Csrs/Cbag: Gainesville, FL, USA, 1994; pp. 56–63. [Google Scholar]
- Woo, P.C.Y.; Tse, H.; Lau, S.K.P.; Leung, K.-W.; Woo, G.K.S.; Wong, M.K.M.; Ho, C.-M.; Yuen, K.-Y. Alkanindiges hongkongensis sp. nov. A novel alkanindiges species isolated from a patient with parotid abscess. Syst. Appl. Microbiol. 2005, 28, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Bogan, B.W.; Sullivan, W.R.; Kayser, K.J.; Derr, K.D.; Aldrich, H.C.; Peterek, J.R. Alkanindiges illinoisensis gen. Nov., sp. nov., an obligately hydrocarbonoclastic, aerobic squalane-degrading bacterium isolated from oilfield soils. Int. J. Syst. Evol. Microbiol. 2003, 53, 1389–1395. [Google Scholar] [CrossRef]
- Li, M.; Lou, H.X.; Huang, C.S.; Lu, Q. Main volatile chemical constituents and antimicrobial activity of scaevola sericea leaves. Technol. Dev. Chem. Ind. 2015, 1, 5. [Google Scholar]
- Prasanna, R.; Gupta, V.; Natarajan, C.; Chaudhary, V. Bioprospecting for genes involved in the production of chitosanases and microcystin-like compounds in anabaena strains. World J. Microbiol. Biotechnol. 2010, 26, 717–724. [Google Scholar] [CrossRef]
- Prasanna, R.; Joshi, M.; Rana, A.; Nain, L. Modulation of iaa production in cyanobacteria by tryptophan and light. Pol. J. Microbiol. 2010, 59, 99–105. [Google Scholar]
- Chaudhary, V.; Prasanna, R.; Bhatnagar, A.K. Modulation of fungicidal potential of anabaena strains by light and temperature. Folia Microbiol. 2012, 57, 199–208. [Google Scholar] [CrossRef]
- Prasanna, R.; Chaudhary, V.; Gupta, V.; Babu, S.; Kumar, A.; Singh, R.; Shivay, Y.S.; Nain, L. Cyanobacteria mediated plant growth promotion and bioprotection against fusarium wilt in tomato. Eur. J. Plant Pathol. 2013, 136, 337–353. [Google Scholar] [CrossRef]
- Santana, R.S.M.; Fernandes, G.W.; Avila, M.P.; Reis, M.P.; de Araujo, F.M.G.; Salim, A.C.M.; Oliveira, G.; Chartone-Souza, E.; Nascimento, A.M.A. Endophytic microbiota associated with the root tips and leaves of baccharis dracunculifolia. Braz. Arch. Biol. Technol. 2016, 59, 11. [Google Scholar] [CrossRef]
- Chakravarthy, S.K.; Ramaprasad, E.V.V.; Shobha, E.; Sasikala, C.; Ramana, C.V. Rhodoplanes piscinae sp nov isolated from pond water. Int. J. Syst. Evol. Microbiol. 2012, 62, 2828–2834. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, A.; Sasikala, C.; Ramana, C.V. Rhodoplanes oryzae sp. nov., a phototrophic alphaproteobacterium isolated from the rhizosphere soil of paddy. Int. J. Syst. Evol. Microbiol. 2014, 64, 2198–2203. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Samples | Shannon | Chao1 | Heip | Coverage |
|---|---|---|---|---|---|
| Fungi | IP | 2.39 ± 0.07c | 279 ± 16b | 0.050 ± 0.001b | 0.9988 ± 0.0001a |
| WC | 1.35 ± 0.14b | 290 ± 21b | 0.013 ± 0.003a | 0.9989 ± 0.0000a | |
| SS | 3.22 ± 0.15d | 483 ± 3c | 0.057 ± 0.005c | 0.9985 ± 0.0003a | |
| CN | 1.33 ± 0.02b | 278 ± 41b | 0.014 ± 0.001a | 0.9988 ± 0.0002a | |
| SP | 0.77 ± 0.15a | 146 ± 44a | 0.013 ± 0.001a | 0.9994 ± 0.0003b | |
| Bacteria | IP | 2.13 ± 0.05a | 498 ± 37a | 0.023 ± 0.001a | 0.9968 ± 0.0008b |
| WC | 4.40 ± 0.02d | 1288 ± 80c | 0.090 ± 0.010c | 0.9911 ± 0.0025ab | |
| SS | 5.52 ± 0.04e | 2070 ± 55e | 0.149 ± 0.011d | 0.9862 ± 0.0030a | |
| CN | 3.87 ± 0.08c | 1549 ± 223d | 0.046 ± 0.005b | 0.9865 ± 0.0036a | |
| SP | 2.78 ± 0.07b | 1034 ± 122b | 0.024 ± 0.003a | 0.9927 ± 0.0016b | |
| Diazotrophs | IP | 1.87 ± 0.20a | 137 ± 16a | 0.045 ± 0.005a | 0.9989 ± 0.0003a |
| WC | 4.30 ± 0.02c | 219 ± 8b | 0.346 ± 0.013d | 0.9992 ± 0.0004a | |
| SS | 3.51 ± 0.25b | 192 ± 30b | 0.183 ± 0.053b | 0.9992 ± 0.0002a | |
| CN | 4.73 ± 0.07d | 467 ± 46d | 0.253 ± 0.044c | 0.9980 ± 0.0006a | |
| SP | 4.33 ± 0.21c | 279 ± 29c | 0.285 ± 0.040cd | 0.9985 ± 0.0008a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bao, L.; Cai, W.; Zhang, X.; Liu, J.; Chen, H.; Wei, Y.; Jia, X.; Bai, Z. Distinct Microbial Community of Phyllosphere Associated with Five Tropical Plants on Yongxing Island, South China Sea. Microorganisms 2019, 7, 525. https://doi.org/10.3390/microorganisms7110525
Bao L, Cai W, Zhang X, Liu J, Chen H, Wei Y, Jia X, Bai Z. Distinct Microbial Community of Phyllosphere Associated with Five Tropical Plants on Yongxing Island, South China Sea. Microorganisms. 2019; 7(11):525. https://doi.org/10.3390/microorganisms7110525
Chicago/Turabian StyleBao, Lijun, Wenyang Cai, Xiaofen Zhang, Jinhong Liu, Hao Chen, Yuansong Wei, Xiuxiu Jia, and Zhihui Bai. 2019. "Distinct Microbial Community of Phyllosphere Associated with Five Tropical Plants on Yongxing Island, South China Sea" Microorganisms 7, no. 11: 525. https://doi.org/10.3390/microorganisms7110525