Deciphering the Infectious Process of Colletotrichum lupini in Lupin through Transcriptomic and Proteomic Analysis

 ,
,  , ,
, ,  and
and
Abstract
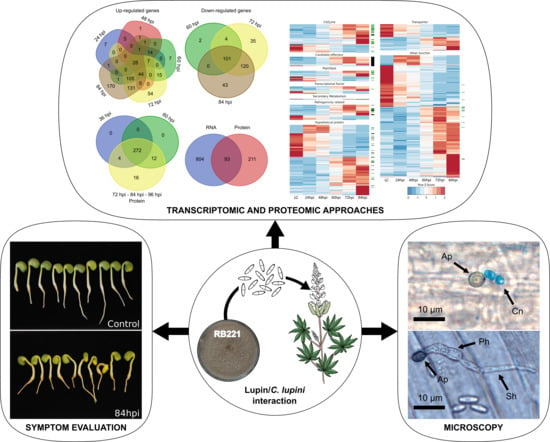
:
1. Introduction
2. Materials and Methods
2.1. Fungal Isolate and Plant Material
2.1.1. Fungal Culture Condition and Inoculum Preparation
2.1.2. Plant Growth
2.1.3. Inoculation and Incubation Conditions
2.2. Preparation of RNA Samples for RNA Sequencing Analysis
2.2.1. Extraction of C. lupini RNA from Pure Culture
2.2.2. Extraction of Total RNA from Inoculated Plants
2.3. Preparation of Libraries, RNA Sequencing and Analysis
2.4. Extraction and Digestion of Total Proteins
2.5. Orbitrap Mass Spectrometry and Analysis
2.6. Transcriptome and Proteome Prediction and Analysis Pipeline
3. Results
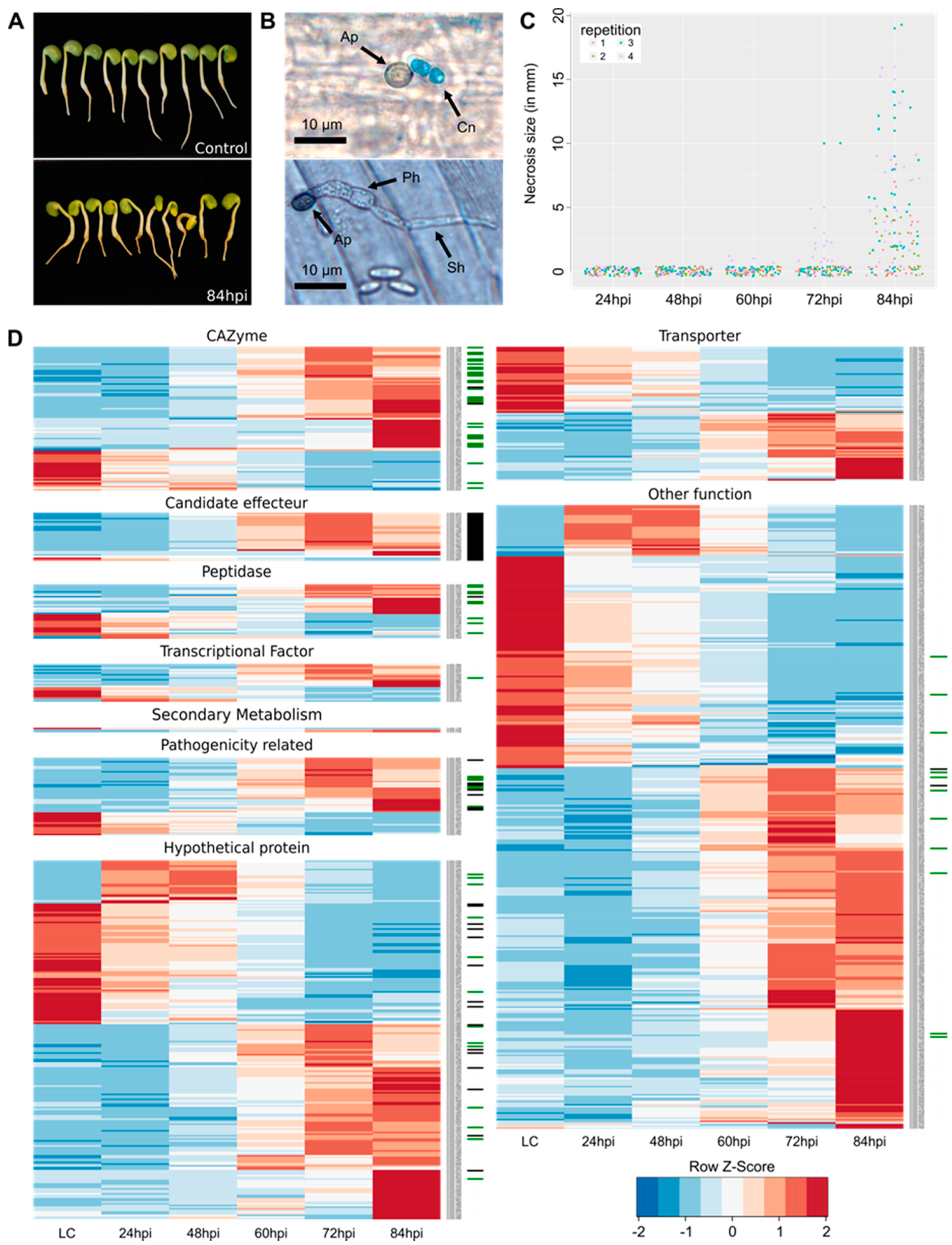
3.1. Symptom Evaluation and Morphological Structures
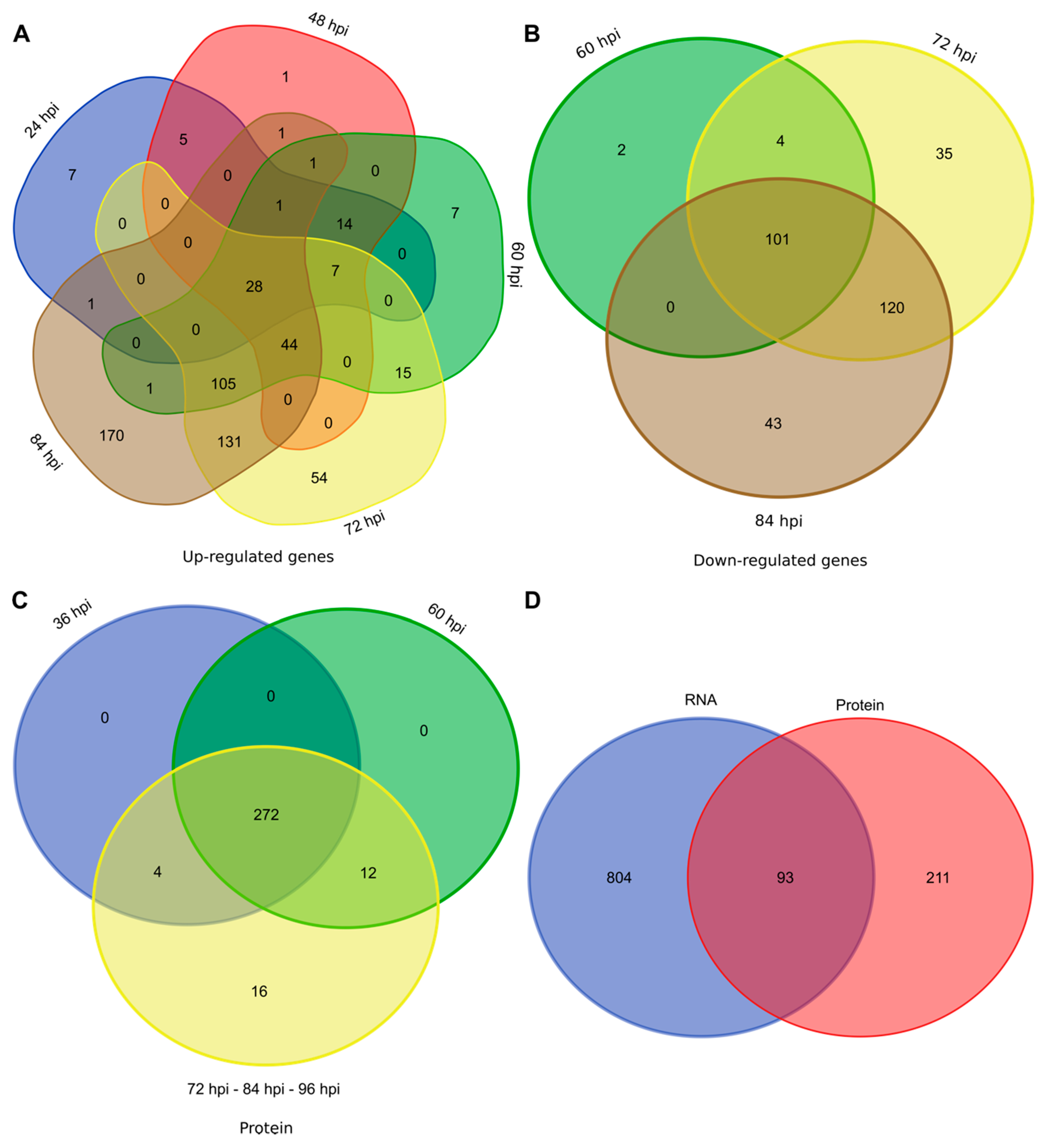
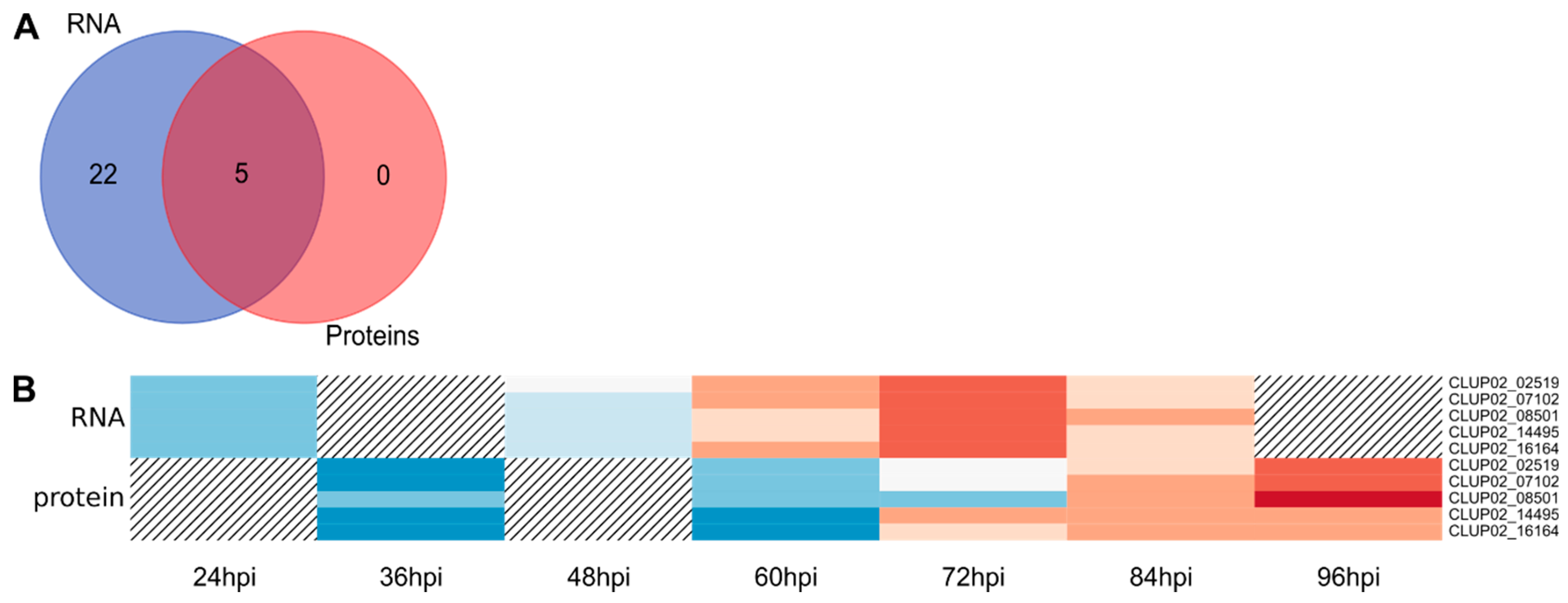
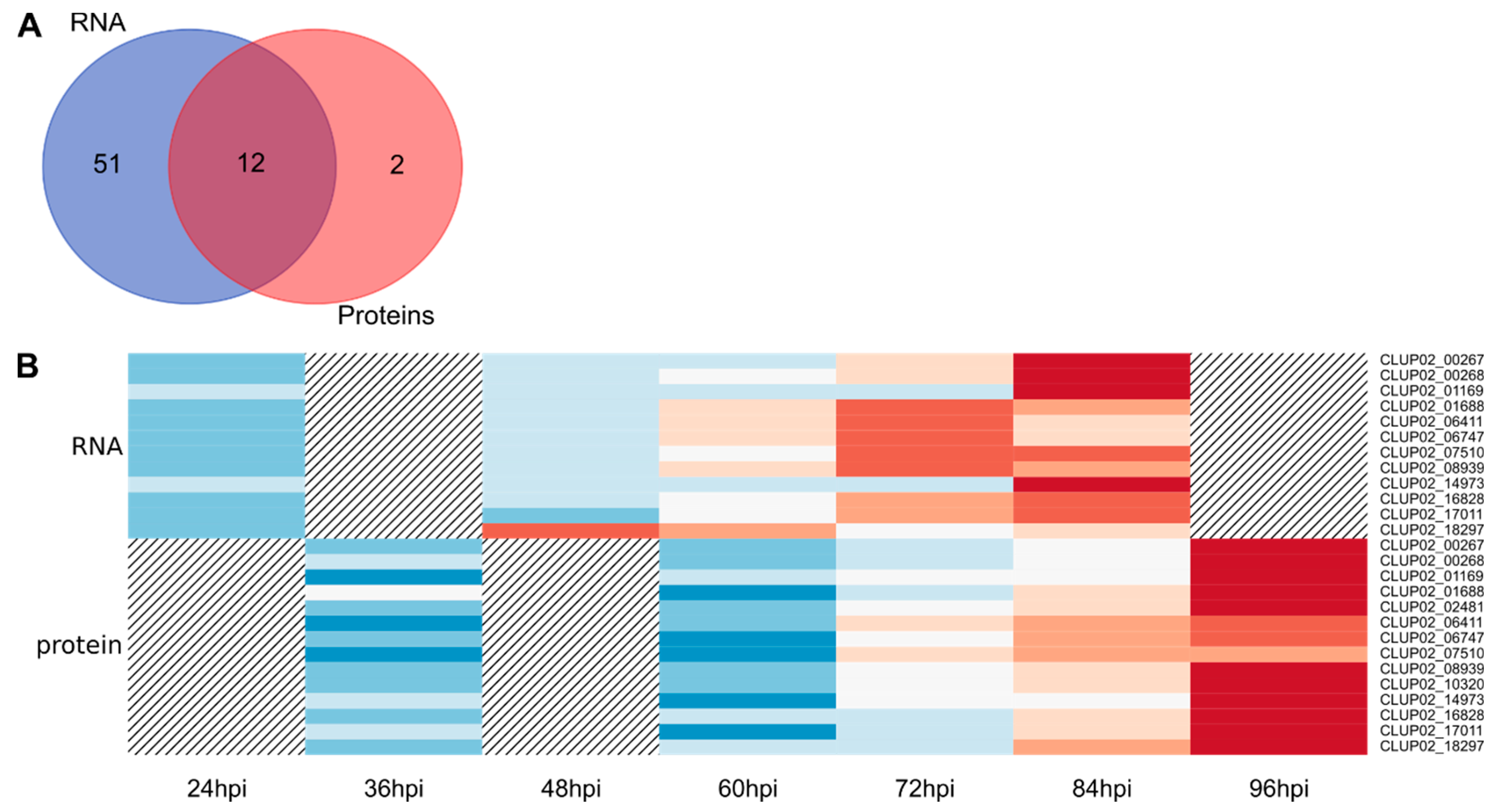
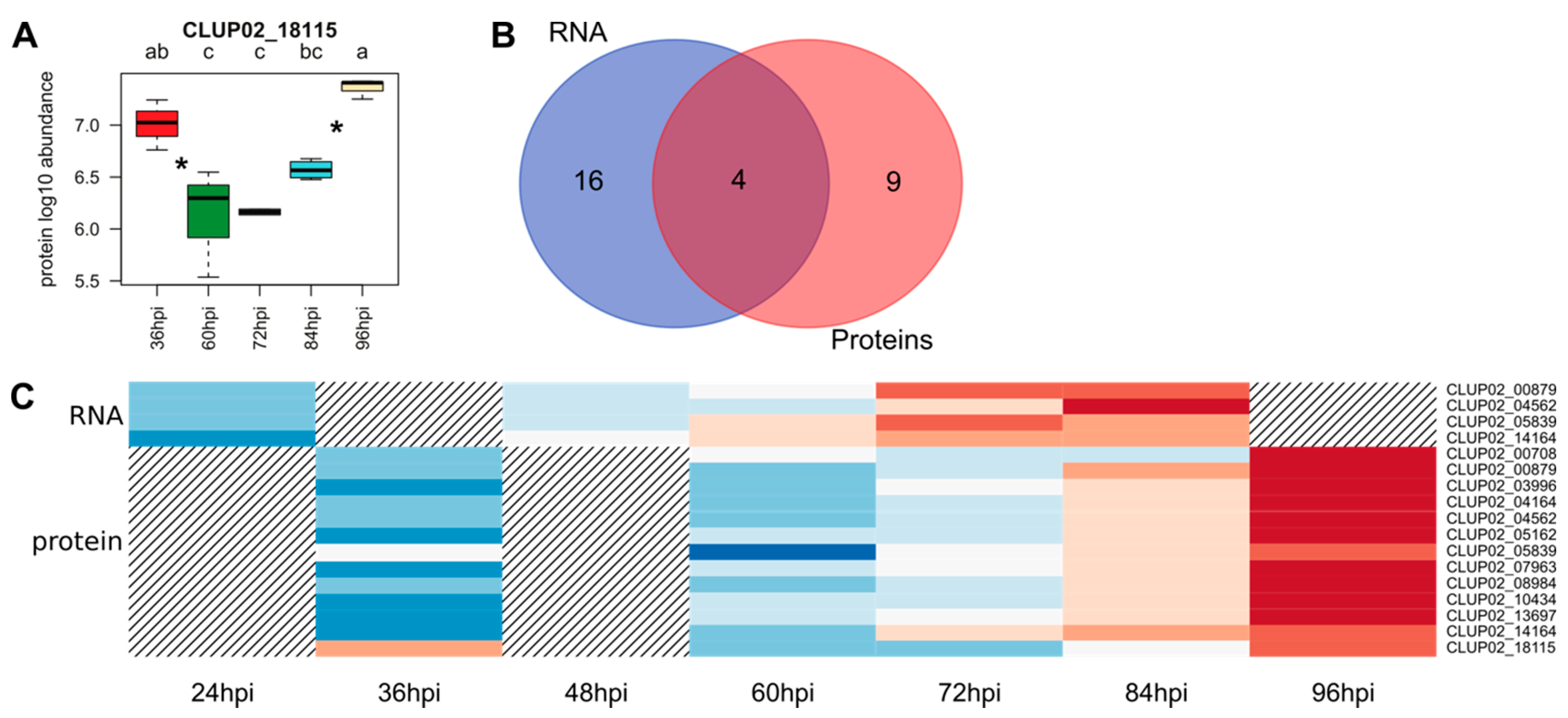
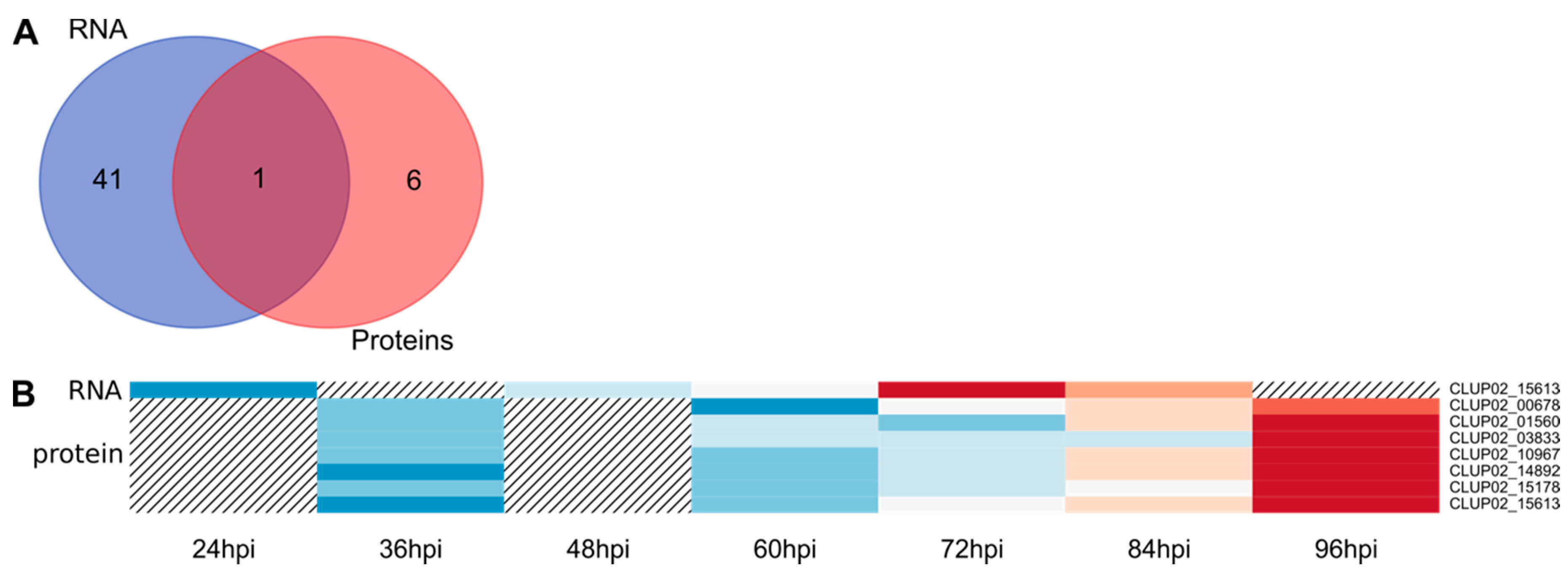
3.2. Transcriptomic and Proteomic Data and Selection
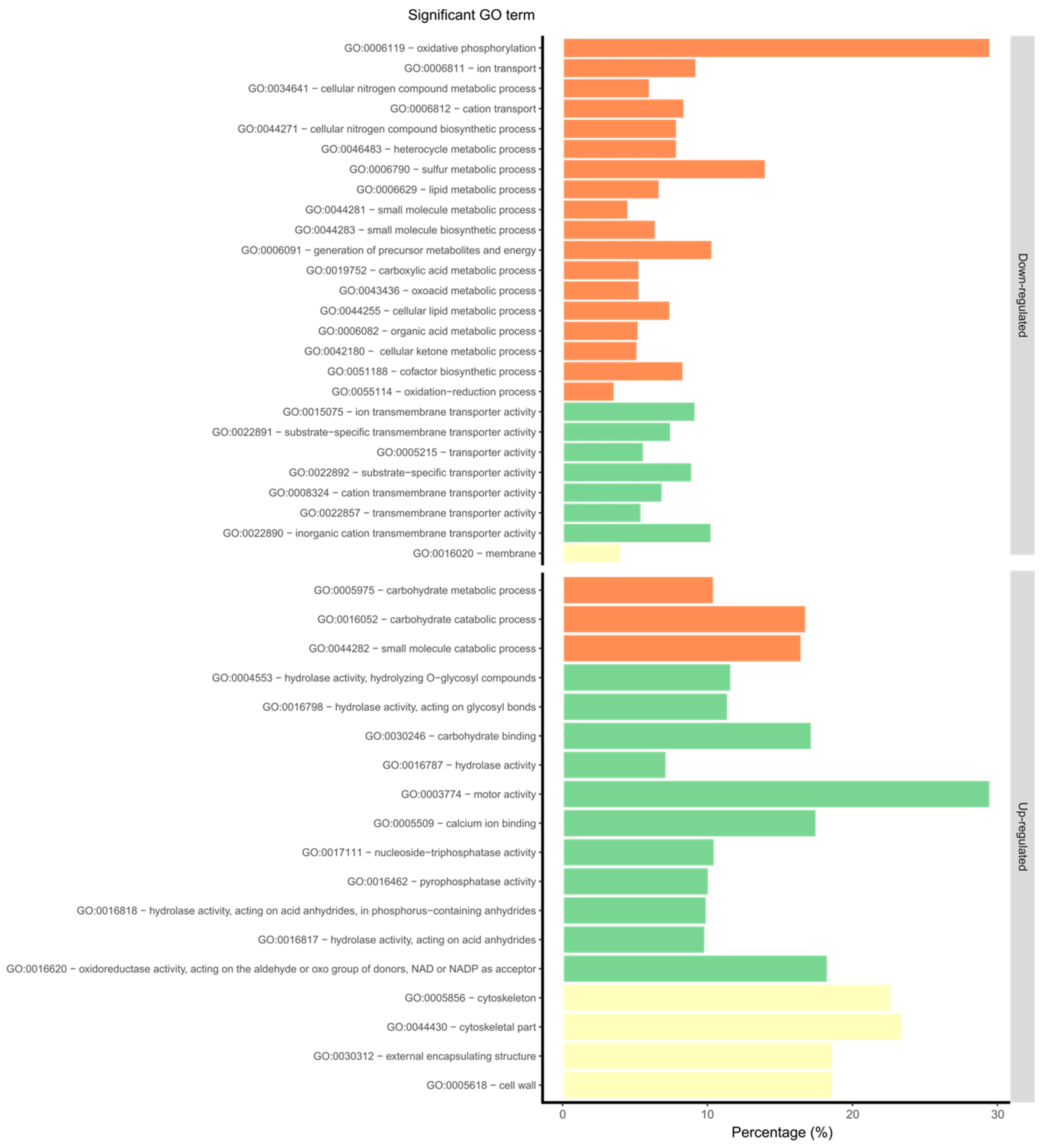
3.3. Overview of Gene Expression and Protein Synthesis
3.4. Expression of the Predicted Functional Class of DEGs and QPs in Lupin Infection
3.4.1. Secreted Proteins, Small-Secreted Peptides (SSP)
3.4.2. Candidate Effectors
3.4.3. Carbohydrate Active Enzymes (CAZymes)
3.4.4. Peptidases
3.4.5. Transmembrane Transporters
3.4.6. Genes Associated with Secondary Metabolism
3.4.7. Transcription Factors
3.4.8. Cytochromes P450
3.4.9. Factors Associated with Pathogenicity and Stress Response
3.4.10. Other Functions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dean, R.; Van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Pietro, A.D.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayawardena, R.S.; Hyde, K.D.; Damm, U.; Cai, L.; Liu, M.; Li, X.H.; Zhang, W.; Zhao, W.; Yan, J. Notes on currently accepted species of Colletotrichum. Mycosphere 2016, 7, 1192–1260. [Google Scholar] [CrossRef]
- Baroncelli, R.; Talhinhas, P.; Pensec, F.; Sukno, S.A.; Le Floch, G.; Thon, M.R. The Colletotrichum acutatum Species Complex as a Model System to Study Evolution and Host Specialization in Plant Pathogens. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gondran, J.; Pacault, D. White lupin anthracnose [Colletotrichum acutatum Simmonds]. Phytoma 1997, 494, 28–31. [Google Scholar]
- Talhinhas, P.; Baroncelli, R.; Le Floch, G. Anthracnose of lupins caused by Colletotrichum lupini: A recent disease and a successful worldwide pathogen. J. Plant Pathol. 2016, 98, 5–14. [Google Scholar]
- Sweetingham, M.; Thomas, G.; Yang, H.; Shea, G. Anthracnose–the pathogen, epidemiology and the management package. In Highlights of Lupin Research and Development in Western Australia; Shea, G., Ed.; The Chief Executive Officer, Agriculture Western Australia: Perth, Australia, 1998; pp. 8–9. [Google Scholar]
- White, P.; French, B.; McLarty, A.; Grains Research and Development Corporation. Producing lupins. In Bulletin 4720; Department of Agriculture and Food: South Perth, Australia, 2008; pp. 1–167. [Google Scholar]
- Gomes, S.; Prieto, P.; Martins-Lopes, P.; Carvalho, T.; Martin, A.; Guedes-Pinto, H. Development of Colletotrichum acutatum on Tolerant and Susceptible Olea europaea L. cultivars: A Microscopic Analysis. Mycopathologia 2009, 168, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Alkan, N.; Friedlander, G.; Ment, D.; Prusky, D.; Fluhr, R. Simultaneous transcriptome analysis of Colletotrichum gloeosporioides and tomato fruit pathosystem reveals novel fungal pathogenicity and fruit defense strategies. New Phytol. 2015, 205, 801–815. [Google Scholar] [CrossRef]
- Kubo, Y.; Takano, Y. Dynamics of infection-related morphogenesis and pathogenesis in Colletotrichum orbiculare. J. Gen. Plant Pathol. 2013, 79, 233–242. [Google Scholar] [CrossRef]
- O’Connell, R.J.; Thon, M.R.; Hacquard, S.; Amyotte, S.G.; Kleemann, J.; Torres, M.F.; Damm, U.; Buiate, E.A.; Epstein, L.; Alkan, N.; et al. Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 2012, 44, 1060–1065. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, X.; He, C.; Zhang, Q.-Y.; Zou, X.; Duan, K.; Gao, Q. Novel Fungal Pathogenicity and Leaf Defense Strategies Are Revealed by Simultaneous Transcriptome Analysis of Colletotrichum fructicola and Strawberry Infected by This Fungus. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Prasanth, C.N.; Viswanathan, R.; Krishna, N.; Malathi, P.; Ramesh Sundar, A.; Tiwari, T. Unraveling the Genetic Complexities in Gene Set of Sugarcane Red Rot Pathogen Colletotrichum falcatum Through Transcriptomic Approach. Sugar Tech 2017, 19, 604–615. [Google Scholar] [CrossRef]
- Kleemann, J.; Rincon-Rivera, L.J.; Takahara, H.; Neumann, U.; van Themaat, E.V.L.; van der Does, H.C.; Hacquard, S.; Stüber, K.; Will, I.; Schmalenbach, W.; et al. Sequential Delivery of Host-Induced Virulence Effectors by Appressoria and Intracellular Hyphae of the Phytopathogen Colletotrichum higginsianum. PLoS Pathog. 2012, 8, e1002643. [Google Scholar] [CrossRef]
- Bhadauria, V.; Vijayan, P.; Wei, Y.; Banniza, S. Transcriptome analysis reveals a complex interplay between resistance and effector genes during the compatible lentil-Colletotrichum lentis interaction. Sci. Rep. 2017, 7, 42338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, S.-A.; Hatfield, J.; Rusu, A.G.; Maclean, D.J.; Manners, J.M. CgDN3: An Essential Pathogenicity Gene of Colletotrichum gloeosporioides Necessary to Avert a Hypersensitive-Like Response in the Host Stylosanthes guianensis. Mol. Plant-Microbe Interact. 2000, 13, 929–941. [Google Scholar] [CrossRef]
- Toruño, T.Y.; Stergiopoulos, I.; Coaker, G. Plant-Pathogen Effectors: Cellular Probes Interfering with Plant Defenses in Spatial and Temporal Manners. Annu. Rev. Phytopathol. 2016, 54, 419–441. [Google Scholar] [CrossRef] [Green Version]
- Gan, P.; Ikeda, K.; Irieda, H.; Narusaka, M.; O’Connell, R.J.; Narusaka, Y.; Takano, Y.; Kubo, Y.; Shirasu, K. Comparative genomic and transcriptomic analyses reveal the hemibiotrophic stage shift of Colletotrichum fungi. New Phytol. 2013, 197, 1236–1249. [Google Scholar] [CrossRef]
- Villa-Rivera, M.G.; Conejo-Saucedo, U.; Lara-Marquez, A.; Cano-Camacho, H.; Lopez-Romero, E.; Zavala-Paramo, M.G. The Role of Virulence Factors in the Pathogenicity of Colletotrichum sp. Curr. Protein Pept. Sci. 2017, 18, 1005–1018. [Google Scholar] [CrossRef]
- Münch, S.; Lingner, U.; Floss, D.S.; Ludwig, N.; Sauer, N.; Deising, H.B. The hemibiotrophic lifestyle of Colletotrichum species. J. Plant Physiol. 2008, 165, 41–51. [Google Scholar] [CrossRef]
- Chagué, V.; Maor, R.; Sharon, A. CgOpt1, a putative oligopeptide transporter from Colletotrichum gloeosporioides that is involved in responses to auxin and pathogenicity. BMC Microbiol. 2009, 9, 173. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yan, Y.; Huang, J.; Hsiang, T.; Wei, Y.; Li, Y.; Gao, J.; Zheng, L. A Novel MFS Transporter Gene ChMfs1 Is Important for Hyphal Morphology, Conidiation, and Pathogenicity in Colletotrichum higginsianum. Front. Microbiol. 2017, 8, 1953. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Park, S.-Y.; Kim, H.; Kim, D.; Lee, S.-W.; Kim, H.T.; Lee, J.-H.; Choi, W. Isolation and Characterization of the Colletotrichum acutatum ABC Transporter CaABC1. Plant Pathol. J. 2014, 30, 375–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pemberton, C.L.; Salmond, G.P.C. The Nep1-like proteins-a growing family of microbial elicitors of plant necrosis. Mol. Plant Pathol. 2004, 5, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.H.; Yarden, O.; Gollop, N.; Chen, S.; Zveibil, A.; Belausov, E.; Freeman, S. Differential protein expression in Colletotrichum acutatum: Changes associated with reactive oxygen species and nitrogen starvation implicated in pathogenicity on strawberry. Mol. Plant Pathol. 2008, 9, 171–190. [Google Scholar] [CrossRef] [PubMed]
- El-Akhal, M.R.; Colby, T.; Cantoral, J.M.; Harzen, A.; Schmidt, J.; Fernández-Acero, F.J. Proteomic analysis of conidia germination in Colletotrichum acutatum. Arch. Microbiol. 2013, 195, 227–246. [Google Scholar] [CrossRef]
- Li, H.; Wu, Y.; Liu, W.; Zhang, X.-M.; Gong, J.-S.; Shi, J.-S.; Xu, Z.-H. iTRAQ-based quantitative proteomic analysis of Colletotrichum lini reveals ethanol induced mechanism for enhancing dihydroxylation efficiency of DHEA. J. Proteom. 2020, 224, 103851. [Google Scholar] [CrossRef]
- Sakaguchi, A.; Miyaji, T.; Tsuji, G.; Kubo, Y. Kelch repeat protein Clakel2p and calcium signaling control appressorium development in Colletotrichum lagenarium. Eukaryotic Cell 2008, 7, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Kodama, S.; Nishiuchi, T.; Kubo, Y. Colletotrichum orbiculare MTF4 Is a Key Transcription Factor Downstream of MOR Essential for Plant Signal-Dependent Appressorium Development and Pathogenesis. Mol. Plant-Microbe Interact. 2018, 32, 313–324. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Tang, J.; Yuan, Q.; Gu, Q.; Liu, H.; Huang, J.; Hsiang, T.; Zheng, L. ChCDC25 Regulates Infection-Related Morphogenesis and Pathogenicity of the Crucifer Anthracnose Fungus Colletotrichum higginsianum. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ren, D.; Guo, H.; Chen, X.; Zhu, P.; Nie, H.; Xu, L. CgSCD1 Is Essential for Melanin Biosynthesis and Pathogenicity of Colletotrichum gloeosporioides. Pathogens 2020, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Huser, A.; Takahara, H.; Schmalenbach, W.; O’Connell, R. Discovery of pathogenicity genes in the crucifer anthracnose fungus Colletotrichum higginsianum, using random insertional mutagenesis. Mol. Plant Microbe Interact. 2009, 22, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhao, D.; Zheng, L.; Hsiang, T.; Wei, Y.; Fu, Y.; Huang, J. Identification of virulence genes in the crucifer anthracnose fungus Colletotrichum higginsianum by insertional mutagenesis. Microb. Pathog. 2013, 64, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Li, G.; Lin, C.; Shi, T.; Zhai, L.; Chen, Y.; Huang, G. Identifying pathogenicity genes in the rubber tree anthracnose fungus Colletotrichum gloeosporioides through random insertional mutagenesis. Microbiol. Res. 2013, 168, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Saubeau, G.; Gaillard, F.; Legentil, L.; Nugier-Chauvin, C.; Ferrières, V.; Andrivon, D.; Val, F. Identification of three elicitins and a galactan-based complex polysaccharide from a concentrated culture filtrate of Phytophthora infestans efficient against Pectobacterium atrosepticum. Molecules 2014, 19, 15374–15390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méchin, V.; Damerval, C.; Zivy, M. Total protein extraction with TCA-acetone. Methods Mol. Biol. 2007, 355, 1–8. [Google Scholar] [CrossRef]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef]
- Langella, O.; Valot, B.; Balliau, T.; Blein-Nicolas, M.; Bonhomme, L.; Zivy, M. X!TandemPipeline: A Tool to Manage Sequence Redundancy for Protein Inference and Phosphosite Identification. J. Proteome Res. 2017, 16, 494–503. [Google Scholar] [CrossRef]
- Valot, B.; Langella, O.; Nano, E.; Zivy, M. MassChroQ: A versatile tool for mass spectrometry quantification. Proteomics 2011, 11, 3572–3577. [Google Scholar] [CrossRef]
- Lyutvinskiy, Y.; Yang, H.; Rutishauser, D.; Zubarev, R.A. In silico instrumental response correction improves precision of label-free proteomics and accuracy of proteomics-based predictive models. Mol. Cell. Proteom. 2013, 12, 2324–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Park, K.-J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Pierleoni, A.; Martelli, P.L.; Casadio, R. PredGPI: A GPI-anchor predictor. BMC Bioinform. 2008, 9, 392. [Google Scholar] [CrossRef] [Green Version]
- Fankhauser, N.; Mäser, P. Identification of GPI anchor attachment signals by a Kohonen self-organizing map. Bioinformatics 2005, 21, 1846–1852. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Benedito, V.A.; Udvardi, M.K.; Zhao, P.X. TransportTP: A two-phase classification approach for membrane transporter prediction and characterization. BMC Bioinform. 2009, 10, 418. [Google Scholar] [CrossRef] [Green Version]
- Khaldi, N.; Seifuddin, F.T.; Turner, G.; Haft, D.; Nierman, W.C.; Wolfe, K.H.; Fedorova, N.D. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet. Biol. 2010, 47, 736–741. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, N.D.; Moktali, V.; Medema, M.H. Bioinformatics approaches and software for detection of secondary metabolic gene clusters. Methods Mol. Biol. 2012, 944, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Baroncelli, R.; Amby, D.B.; Zapparata, A.; Sarrocco, S.; Vannacci, G.; Le Floch, G.; Harrison, R.J.; Holub, E.; Sukno, S.A.; Sreenivasaprasad, S.; et al. Gene family expansions and contractions are associated with host range in plant pathogens of the genus Colletotrichum. BMC Genom. 2016, 17, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi-Derazmahalleh, M.; Chang, S.; Thomas, G.; Derbyshire, M.; Bayer, P.E.; Edwards, D.; Nelson, M.N.; Erskine, W.; Lopez-Ruiz, F.J.; Clements, J.; et al. Prediction of pathogenicity genes involved in adaptation to a lupin host in the fungal pathogens Botrytis cinerea and Sclerotinia sclerotiorum via comparative genomics. BMC Genom. 2019, 20, 385. [Google Scholar] [CrossRef] [PubMed]
- Bhadauria, V.; Banniza, S.; Vandenberg, A.; Selvaraj, G.; Wei, Y. Overexpression of a Novel Biotrophy-Specific Colletotrichum truncatum Effector, CtNUDIX, in Hemibiotrophic Fungal Phytopathogens Causes Incompatibility with Their Host Plants. Eukaryot. Cell 2013, 12, 2–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]
- Schmidt, S.M.; Houterman, P.M.; Schreiver, I.; Ma, L.; Amyotte, S.; Chellappan, B.; Boeren, S.; Takken, F.L.W.; Rep, M. MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genom. 2013, 14, 119. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, B.; Várnai, A.; Røhr, Å.K.; Eijsink, V.G.H. Oxidoreductases and Reactive Oxygen Species in Conversion of Lignocellulosic Biomass. Microbiol. Mol. Biol. Rev. 2018, 82, e00029-18. [Google Scholar] [CrossRef] [Green Version]
- Pilgaard, B.; Wilkens, C.; Herbst, F.-A.; Vuillemin, M.; Rhein-Knudsen, N.; Meyer, A.S.; Lange, L. Proteomic enzyme analysis of the marine fungus Paradendryphiella salina reveals alginate lyase as a minimal adaptation strategy for brown algae degradation. Sci. Rep. 2019, 9, 12338. [Google Scholar] [CrossRef] [Green Version]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; de Vries, R.P.; Mäkelä, M.R. Plant-Polysaccharide-Degrading Enzymes from Basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-X.; Yendrek, C.R.; Caetano-Anolles, G.; Hartman, G.L. Genomic characterization of plant cell wall degrading enzymes and in silico analysis of xylanses and polygalacturonases of Fusarium virguliforme. BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liu, H.; Wang, C.; Xu, J.-R. Correction: Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genom. 2014, 15, 6. [Google Scholar] [CrossRef] [Green Version]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.L.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic Analysis of the Necrotrophic Fungal Pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [Green Version]
- Glass, N.L.; Schmoll, M.; Cate, J.H.D.; Coradetti, S. Plant Cell Wall Deconstruction by Ascomycete Fungi. Annu. Rev. Microbiol. 2013, 67, 477–498. [Google Scholar] [CrossRef]
- Eley, K.L.; Halo, L.M.; Song, Z.; Powles, H.; Cox, R.J.; Bailey, A.M.; Lazarus, C.M.; Simpson, T.J. Biosynthesis of the 2-Pyridone Tenellin in the Insect Pathogenic Fungus Beauveria bassiana. ChemBioChem 2007, 8, 289–297. [Google Scholar] [CrossRef]
- Cary, J.W.; Uka, V.; Han, Z.; Buyst, D.; Harris-Coward, P.Y.; Ehrlich, K.C.; Wei, Q.; Bhatnagar, D.; Dowd, P.F.; Martens, S.L.; et al. An Aspergillus flavus secondary metabolic gene cluster containing a hybrid PKS-NRPS is necessary for synthesis of the 2-pyridones, leporins. Fungal Genet. Biol. 2015, 81, 88–97. [Google Scholar] [CrossRef]
- Qiao, K.; Chooi, Y.-H.; Tang, Y. Identification and engineering of the cytochalasin gene cluster from Aspergillus clavatus NRRL 1. Metab. Eng. 2011, 13, 723–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qutob, D.; Kemmerling, B.; Brunner, F.; Küfner, I.; Engelhardt, S.; Gust, A.A.; Luberacki, B.; Seitz, H.U.; Stahl, D.; Rauhut, T.; et al. Phytotoxicity and Innate Immune Responses Induced by Nep1-Like Proteins. Plant Cell 2006, 18, 3721–3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perfect, S.E.; O’Connell, R.J.; Green, E.F.; Doering-Saad, C.; Green, J.R. Expression cloning of a fungal proline-rich glycoprotein specific to the biotrophic interface formed in the Colletotrichum–bean interaction. Plant J. 1998, 15, 273–279. [Google Scholar] [CrossRef]
- Takahara, H.; Hacquard, S.; Kombrink, A.; Hughes, H.B.; Halder, V.; Robin, G.P.; Hiruma, K.; Neumann, U.; Shinya, T.; Kombrink, E.; et al. Colletotrichum higginsianum extracellular LysM proteins play dual roles in appressorial function and suppression of chitin-triggered plant immunity. New Phytol. 2016, 211, 1323–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottmann, C.; Luberacki, B.; Küfner, I.; Koch, W.; Brunner, F.; Weyand, M.; Mattinen, L.; Pirhonen, M.; Anderluh, G.; Seitz, H.U.; et al. A common toxin fold mediates microbial attack and plant defense. Proc. Natl. Acad. Sci. USA 2009, 106, 10359–10364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirenberg, H.I.; Feiler, U.; Hagedorn, G. Description of Colletotrichum lupini comb. nov. in modern terms. Mycologia 2002, 94, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Talhinhas, P.; Sreenivasaprasad, S.; Neves-Martins, J.; Oliveira, H. Genetic and Morphological Characterization of Colletotrichum acutatum Causing Anthracnose of Lupins. Phytopathology 2002, 92, 986–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damm, U.; Cannon, P.F.; Woudenberg, J.H.C.; Crous, P.W. The Colletotrichum acutatum species complex. Stud. Mycol. 2012, 73, 37–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riegel, R.; Véliz, D.; von Baer, I.; Quitral, Y.; Muñoz, M. Genetic diversity and virulence of Colletotrichum lupini isolates collected in Chile. Trop. Plant Pathol. 2010, 35, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Dubrulle, G.; Pensec, F.; Picot, A.; Rigalma, K.; Pawtowski, A.; Nicolleau, S.; Harzic, N.; Nodet, P.; Baroncelli, R.; Le Floch, G. Phylogenetic Diversity and Effect of Temperature on Pathogenicity of Colletotrichum lupini. Plant Dis. 2020, 104, 938–950. [Google Scholar] [CrossRef]
- Thomas, G.J.; Adcock, K.G. Exposure to dry heat reduces anthracnose infection of lupin seed. Australas. Plant Pathol. 2004, 33, 537–540. [Google Scholar] [CrossRef]
- Thomas, G.J.; Sweetingham, M.W.; Yang, H.A.; Speijers, J. Effect of temperature on growth of Colletotrichum lupini and on anthracnose infection and resistance in lupins. Australas. Plant Pathol. 2008, 37, 35–39. [Google Scholar] [CrossRef]
- Kubo, Y. Appressorium Function in Colletotrichum orbiculare and Prospect for Genome Based Analysis. In Morphogenesis and Pathogenicity in Fungi; Pérez-Martín, J., Di Pietro, A., Eds.; Topics in Current Genetics; Springer: Berlin/Heidelberg, Germany, 2012; pp. 115–131. ISBN 978-3-642-22916-9. [Google Scholar]
- Lin, C.; Liu, X.; Shi, T.; Li, C.; Huang, G. The Colletotrichum gloeosporioides perilipin homologue CAP 20 regulates functional appressorial formation and fungal virulence. J. Phytopathol. 2018, 166, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Zampounis, A.; Pigné, S.; Dallery, J.-F.; Wittenberg, A.H.J.; Zhou, S.; Schwartz, D.C.; Thon, M.R.; O’Connell, R.J. Genome Sequence and Annotation of Colletotrichum higginsianum, a Causal Agent of Crucifer Anthracnose Disease. Genome Announc. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Wösten, H.A. Hydrophobins: Multipurpose proteins. Annu. Rev. Microbiol. 2001, 55, 625–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.-N.; Wu, Q.-Y.; Zhang, G.-Z.; Zhu, Y.-Y.; Murphy, R.W.; Liu, Z.; Zou, C.-G. Systematic analyses reveal uniqueness and origin of the CFEM domain in fungi. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeZwaan, T.M.; Carroll, A.M.; Valent, B.; Sweigard, J.A. Magnaporthe grisea pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell 1999, 11, 2013–2030. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, R.D.; Kelkar, H.S.; Dean, R.A. An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem. Sci. 2003, 28, 118–121. [Google Scholar] [CrossRef]
- Sygmund, C.; Klausberger, M.; Felice, A.K.; Ludwig, R. Reduction of quinones and phenoxy radicals by extracellular glucose dehydrogenase from Glomerella cingulata suggests a role in plant pathogenicity. Microbiology 2011, 157, 3203–3212. [Google Scholar] [CrossRef] [PubMed]
- Ciuffetti, L.M.; Manning, V.A.; Pandelova, I.; Faris, J.D.; Friesen, T.L.; Strelkov, S.E.; Weber, G.L.; Goodwin, S.B.; Wolpert, T.J.; Figueroa, M. Pyrenophora tritici-repentis: A plant pathogenic fungus with global impact. Genom. Plant-Assoc. Fungi Monocot Pathog. 2014, 1–39. [Google Scholar] [CrossRef]
- De Jonge, R.; Thomma, B.P.H.J. Fungal LysM effectors: Extinguishers of host immunity? Trends Microbiol. 2009, 17, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Whiteford, J.R.; Spanu, P.D. The Hydrophobin HCf-1 of Cladosporium fulvum Is Required for Efficient Water-Mediated Dispersal of Conidia. Fungal Genet. Biol. 2001, 32, 159–168. [Google Scholar] [CrossRef]
- Nyarko, A.; Singarapu, K.K.; Figueroa, M.; Manning, V.A.; Pandelova, I.; Wolpert, T.J.; Ciuffetti, L.M.; Barbar, E. Solution NMR Structures of Pyrenophora tritici-repentis ToxB and Its Inactive Homolog Reveal Potential Determinants of Toxin Activity. J. Biol. Chem. 2014, 289, 25946–25956. [Google Scholar] [CrossRef] [Green Version]
- Sperschneider, J.; Gardiner, D.M.; Dodds, P.N.; Tini, F.; Covarelli, L.; Singh, K.B.; Manners, J.M.; Taylor, J.M. EffectorP: Predicting fungal effector proteins from secretomes using machine learning. New Phytol. 2016, 210, 743–761. [Google Scholar] [CrossRef] [Green Version]
- Laurent, B.; Moinard, M.; Spataro, C.; Ponts, N.; Barreau, C.; Foulongne-Oriol, M. Landscape of genomic diversity and host adaptation in Fusarium graminearum. BMC Genom. 2017, 18, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Sorbo, G.; Schoonbeek, H.; De Waard, M.A. Fungal transporters involved in efflux of natural toxic compounds and fungicides. Fungal Genet. Biol. 2000, 30, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Stefanato, F.L.; Abou-Mansour, E.; Buchala, A.; Kretschmer, M.; Mosbach, A.; Hahn, M.; Bochet, C.G.; Métraux, J.-P.; Schoonbeek, H. The ABC transporter BcatrB from Botrytis cinerea exports camalexin and is a virulence factor on Arabidopsis thaliana. Plant J. 2009, 58, 499–510. [Google Scholar] [CrossRef]
- Pereira, M.F.; de Araújo Dos Santos, C.M.; de Araújo, E.F.; de Queiroz, M.V.; Bazzolli, D.M.S. Beginning to understand the role of sugar carriers in Colletotrichum lindemuthianum: The function of the gene mfs1. J. Microbiol. 2013, 51, 70–81. [Google Scholar] [CrossRef]
- Mayer, A.M.; Staples, R.C.; Gil-ad, N.L. Mechanisms of survival of necrotrophic fungal plant pathogens in hosts expressing the hypersensitive response. Phytochemistry 2001, 58, 33–41. [Google Scholar] [CrossRef]
- Torres, M.A. ROS in biotic interactions. Physiol. Plant 2010, 138, 414–429. [Google Scholar] [CrossRef]
- Goodwin, P.H.; Li, J.; Jin, S. A catalase gene of Colletotrichum gloeosporioides f. sp. malvae is highly expressed during the necrotrophic phase of infection of round-leaved mallow, Malva pusilla. FEMS Microbiol. Lett. 2001, 202, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Pazzagli, L.; Seidl-Seiboth, V.; Barsottini, M.; Vargas, W.A.; Scala, A.; Mukherjee, P.K. Cerato-platanins: Elicitors and effectors. Plant Sci. 2014, 228, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Bhadauria, V.; Banniza, S.; Vandenberg, A.; Selvaraj, G.; Wei, Y. EST mining identifies proteins putatively secreted by the anthracnose pathogen Colletotrichum truncatum. BMC Genom. 2011, 12, 327. [Google Scholar] [CrossRef] [PubMed]
- Ashwin, N.M.R.; Barnabas, L.; Ramesh Sundar, A.; Malathi, P.; Viswanathan, R.; Masi, A.; Agrawal, G.K.; Rakwal, R. Comparative secretome analysis of Colletotrichum falcatum identifies a cerato-platanin protein (EPL1) as a potential pathogen-associated molecular pattern (PAMP) inducing systemic resistance in sugarcane. J. Proteom. 2017, 169, 2–20. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
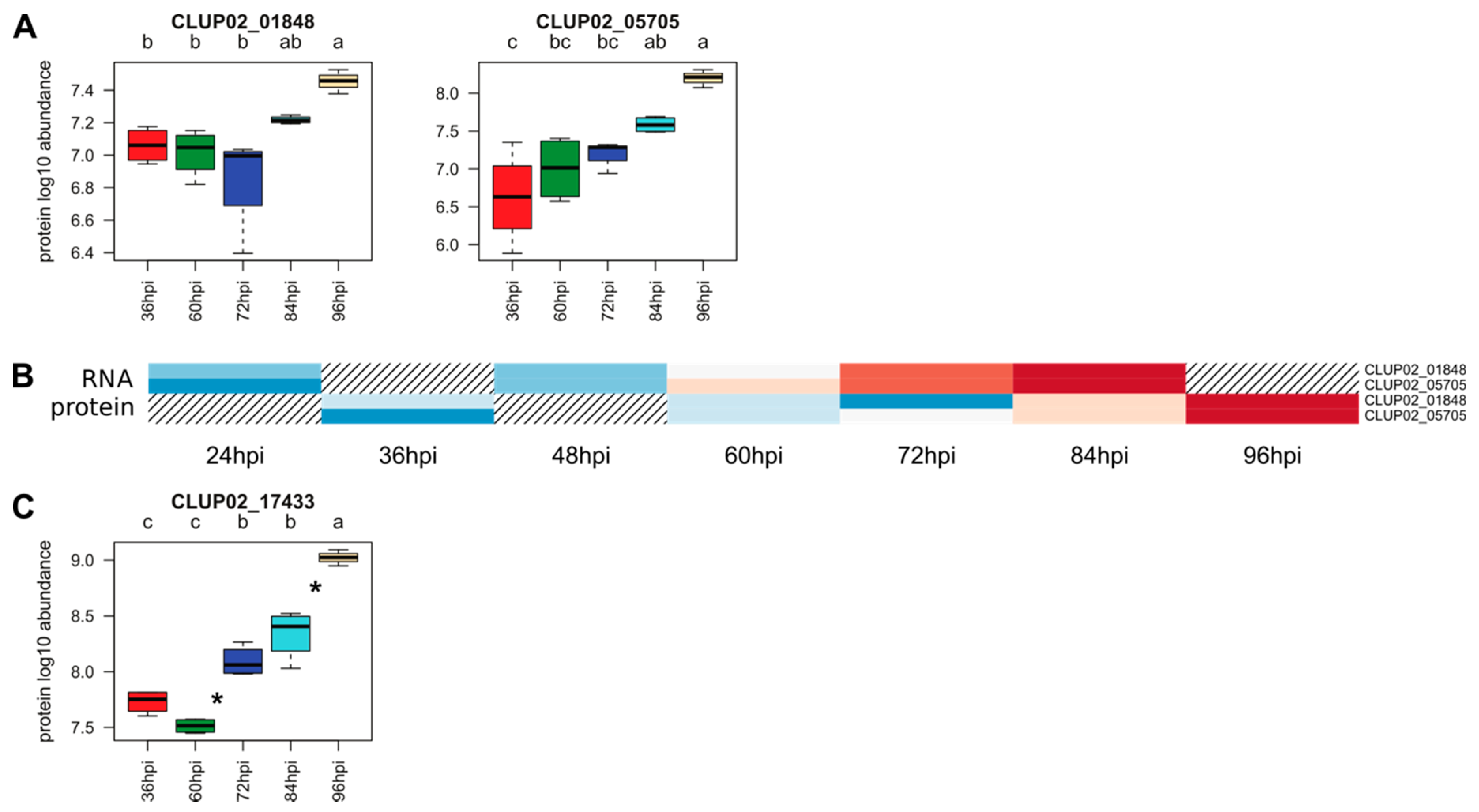{kind=link}
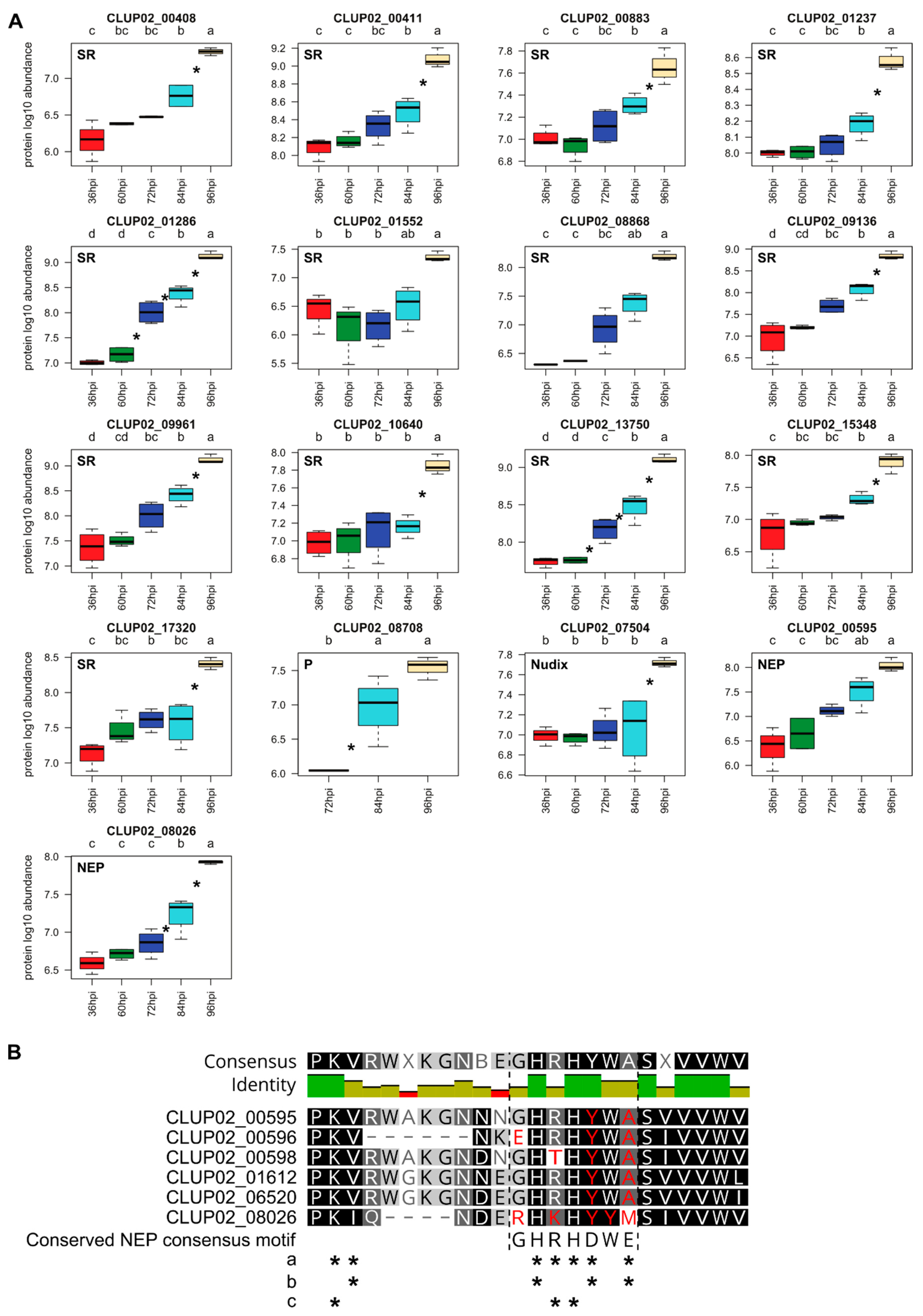{kind=link}
| Transcriptomic Analysis | Proteomic Analysis | Shared between Methods | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 hpi | 48 hpi | 60 hpi | 72 hpi | 84 hpi | Total Number of Transcripts | 36 hpi | 60 hpi | 72/84/96 hpi | Total Number of Proteins | ||
| Protein predicted with signal peptide—SignalP 5.0 | 9 | 37 | 72 | 110 | 119 | 135 | 37 | 37 | 40 | 40 | 31 |
| Extracellular protein—WoLF PSORT | 10 | 37 | 79 | 115 | 130 | 144 | 35 | 35 | 38 | 38 | 31 |
| Transmembrane protein—TMHMM | 14 | 15 | 40 | 77 | 86 | 118 | 21 | 21 | 21 | 21 | 6 |
| Protein containing GPI anchor—PredGPI and GPI-som | 3 | 3 | 9 | 13 | 18 | 21 | 8 | 8 | 9 | 9 | 3 |
| Secreted protein (Extr, Signal P, no TMH, no GPI) | 5 | 32 | 60 | 84 | 96 | 103 | 25 | 25 | 27 | 27 | 26 |
| Small-secreted protein (secreted protein with size < 300 Amino Acid residues) | 2 | 17 | 30 | 41 | 43 | 46 | 6 | 6 | 8 | 8 | 8 |
| Total of DEG or QP at each time point | 63 | 102 | 223 | 384 | 483 | 593 | 276 | 284 | 304 | 304 | 93 |
| Functional Class | Family | 24 hpi | 48 hpi | 60 hpi | 72 hpi | 84 hpi | Total |
|---|---|---|---|---|---|---|---|
| Carbohydrate active enzyme | Auxiliary Activities * | 0 | 1 | 5 | 7 | 13 | 13 |
| Glycoside hydrolases | 0 | 6 | 11 | 22 | 33 | 37 | |
| Carbohydrate binding modules | 0 | 0 | 0 | 0 | 2 | 2 | |
| Glycosyl Transferases | 0 | 0 | 1 | 3 | 6 | 6 | |
| Carbohydrate Esterases | 0 | 3 | 6 | 6 | 6 | 6 | |
| Polysaccharide Lyases | 0 | 0 | 1 | 1 | 1 | 1 | |
| Total of CAZyme domains | 0 | 10 | 24 | 39 | 61 | 65 | |
| Total of genes encoding CAZymes | 0 | 9 | 23 | 38 | 59 | 63 | |
| Transcription factor | Fungal Zn(2)-Cys(6) binuclear cluster domain | 0 | 0 | 2 | 2 | 4 | 4 |
| Fungal specific transcription factor | 0 | 0 | 2 | 4 | 6 | 6 | |
| Helix-loop-helix DNA-binding domain | 0 | 0 | 0 | 0 | 1 | 1 | |
| Zinc finger, C2H2 type | 1 | 1 | 2 | 3 | 2 | 4 | |
| bZIP transcription factor | 0 | 0 | 0 | 1 | 2 | 2 | |
| Spt20 family | 1 | 0 | 0 | 0 | 0 | 1 | |
| CP2 transcription factor | 0 | 0 | 0 | 1 | 0 | 1 | |
| Total of transcription factor domain | 2 | 1 | 6 | 11 | 15 | 19 | |
| Total of genes encoding transcription factors | 2 | 1 | 4 | 9 | 12 | 16 | |
| Secondary metabolism | Polyketide synthase dehydratase | 0 | 0 | 1 | 2 | 2 | 2 |
| Condensation domain | 0 | 0 | 1 | 2 | 2 | 2 | |
| Phosphopantetheine attachment site | 0 | 0 | 1 | 2 | 2 | 2 | |
| Ketoreductase domain (KR) | 0 | 0 | 1 | 2 | 2 | 2 | |
| Total of secondary metabolism domains | 0 | 0 | 4 | 8 | 8 | 8 | |
| Total of secondary metabolism associated genes | 0 | 0 | 2 | 2 | 2 | 2 | |
| Candidate effector | 1 | 11 | 19 | 26 | 24 | 27 | |
| Peptidase | Serine peptidase | 1 | 2 | 3 | 6 | 9 | 10 |
| Metallopeptidase | 0 | 1 | 2 | 3 | 6 | 6 | |
| Cystein peptidase | 2 | 1 | 1 | 0 | 3 | 3 | |
| Peptidase inhibitor | 0 | 0 | 0 | 0 | 1 | 1 | |
| Total of genes encoding peptidases | 3 | 4 | 6 | 9 | 19 | 20 | |
| Transmembrane transporter | The Ca2+:Cation Antiporter (CaCA) Family | 0 | 0 | 1 | 4 | 0 | 4 |
| The Amino Acid/Auxin Permease (AAAP) Family | 0 | 0 | 0 | 3 | 2 | 3 | |
| The ATP-binding Cassette (ABC) Superfamily | 0 | 1 | 2 | 4 | 4 | 5 | |
| The Major Facilitator Superfamily (MFS) | 0 | 0 | 3 | 7 | 13 | 14 | |
| The Amino Acid-Polyamine-Organocation (APC) Family | 0 | 0 | 3 | 2 | 3 | 4 | |
| The Proton-dependent Oligopeptide Transporter (POT) Family | 0 | 0 | 0 | 0 | 1 | 1 | |
| The Small Conductance Mechanosensitive Ion Channel (MscS) Family | 0 | 0 | 0 | 1 | 1 | 1 | |
| The P-type ATPase (P-ATPase) Superfamily | 0 | 0 | 1 | 3 | 2 | 4 | |
| The Mitochondrial Carrier (MC) Family | 0 | 0 | 1 | 1 | 1 | 1 | |
| The Oligopeptide Transporter (OPT) Family | 0 | 0 | 0 | 0 | 1 | 1 | |
| The Transient Receptor Potential Ca2+ Channel (TRP-CC) Family | 0 | 0 | 0 | 1 | 1 | 1 | |
| Threonine/serine exporter | 0 | 0 | 0 | 1 | 1 | 1 | |
| Mitochondrial calcium uniporter | 0 | 0 | 0 | 0 | 1 | 1 | |
| Total of transmembrane transporter genes | 0 | 1 | 11 | 27 | 31 | 41 | |
| Other pathogenicity related-factors | Nudix | 0 | 0 | 1 | 1 | 2 | 2 |
| Necrosis and ethylene-inducing peptide * | 0 | 3 | 6 | 6 | 6 | 6 | |
| Stress response | 0 | 0 | 3 | 12 | 13 | 16 | |
| Pathogenicity | 0 | 2 | 3 | 4 | 8 | 9 | |
| Oxydoreduction | Cytochrome P450 | 0 | 0 | 2 | 7 | 10 | 11 |
| NAD/FAD binding and other oxydo-reductase | 3 | 8 | 17 | 37 | 53 | 58 | |
| Cellular process | AAA family | 0 | 0 | 0 | 0 | 2 | 2 |
| Binding | 6 | 7 | 14 | 15 | 10 | 24 | |
| Cell differentiation | 1 | 1 | 1 | 1 | 0 | 1 | |
| DNA repair | 1 | 1 | 1 | 1 | 1 | 1 | |
| Intracellular transport | 1 | 1 | 3 | 10 | 8 | 13 | |
| Methyltransferase | 1 | 1 | 3 | 6 | 10 | 13 | |
| Mitochondria | 1 | 1 | 2 | 2 | 1 | 2 | |
| Protein kinase | 1 | 1 | 2 | 3 | 6 | 7 | |
| Transcription | 4 | 3 | 3 | 6 | 4 | 8 | |
| Translation | 3 | 5 | 9 | 10 | 5 | 12 | |
| Ubiquitinilation | 1 | 1 | 1 | 2 | 4 | 4 | |
| Metabolic process | Alpha/beta hydrolase | 0 | 0 | 1 | 1 | 2 | 2 |
| AMP-binding enzyme | 0 | 0 | 1 | 1 | 2 | 2 | |
| Carbohydrate metabolism | 1 | 1 | 2 | 2 | 6 | 6 | |
| Coenzyme A metabolism | 0 | 0 | 0 | 0 | 1 | 1 | |
| Lipid metabolism | 0 | 0 | 0 | 4 | 5 | 6 | |
| Cell structure | 4 | 4 | 6 | 9 | 6 | 11 | |
| Hypothetical protein | 27 | 33 | 60 | 93 | 120 | 144 | |
| Others | 2 | 3 | 18 | 40 | 51 | 63 | |
| Substrate | Number of Enzyme | CAZymes | Target | References | ||||
|---|---|---|---|---|---|---|---|---|
| Plant Cell Wall Degrading Enzyme | Fungal Cell Wall Degrading Enzyme | Energy Storage and Recovery | Protein Glycosylation | Fungal Cell Wall Synthesizing Enzyme | ||||
| Cellulose | 1 | AA3_1 | X | [60,61,62] | ||||
| 8 | AA9 | X | [60,61,62,63] | |||||
| 1 | GH7 | X | [11,60,62,63,64,65,66] | |||||
| Hemicellulose (Xyloglucan or Mannogalactan) | 4 | GH31 | X | X | X | [11,60,61,62,63,64,65,66] | ||
| 2 | GH35 | X | [11,60,61,62,63,64,65,66] | |||||
| Pectin | 3 | GH28 | X | [11,60,61,62,63,64,65,66] | ||||
| 1 | GH88/GH105 | X | [11,61,62,63,64,65] | |||||
| 2 | CE8 | X | [11,61,62,63,65,66] | |||||
| 1 | PL1_4 | X | [11,61,62,63,64,65,66] | |||||
| Lignin | 1 | AA1 | X | [60] | ||||
| 1 | AA3 | X | [60] | |||||
| 1 | AA3_2 | X | [60] | |||||
| 1 | AA3_3 | X | [60] | |||||
| Starch | 1 | CBM20 | [63] | |||||
| 1 | GH15 | X | [61,63,64,65] | |||||
| Hemicellulose or Pectin | 1 | CE12 | X | [11,60,61,62,63,65,66] | ||||
| Hemicellulose (Xylan) or Chitin | 1 | CE4 | X | [61,63,65] | ||||
| 2 | GH2 | X | X | [60,61,62,63,64,65,66] | ||||
| 4 | GH3 | X | X | [60,61,62,63,64,65,66] | ||||
| Chitin or Cellulose | 2 | GH1 | X | X | [60,61,62,63,64,65,66] | |||
| 1 | GH5 | X | X | [60,61,62,63,64,65,66] | ||||
| 1 | CBM33/AA10 | X | [60,66] | |||||
| Chitin | 4 | GH18 | X | [61,63,64,65] | ||||
| 1 | GH76 | X | [61,64,65] | |||||
| 1 | GT2 | X | [61,63] | |||||
| 2 | GT2_Chitin_synth_2 | X | [61,63] | |||||
| Glucan (β-1,3-glucan) | 1 | GH131 | X | [61,63] | ||||
| 1 | GH17 | X | [61,63,65] | |||||
| Glycoproteins (N-/O-glucans) | 1 | GH63 | X | [61,64] | ||||
| 1 | GH47 | X | [61,64] | |||||
| Trehalose | 1 | GH37 | X | [61,63,64] | ||||
| Glycogen or Starch | 1 | GT35 | X | [63] | ||||
| - | 1 | GH92 | X | [65] | ||||
| 1 | GT1 | - | ||||||
| 2 | GT2_Glyco_tranf_2_3 | - | ||||||
| 1 | GH30 | X | [61,64] | |||||
| 2 | CE10 | - | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubrulle, G.; Picot, A.; Madec, S.; Corre, E.; Pawtowski, A.; Baroncelli, R.; Zivy, M.; Balliau, T.; Le Floch, G.; Pensec, F. Deciphering the Infectious Process of Colletotrichum lupini in Lupin through Transcriptomic and Proteomic Analysis. Microorganisms 2020, 8, 1621. https://doi.org/10.3390/microorganisms8101621
Dubrulle G, Picot A, Madec S, Corre E, Pawtowski A, Baroncelli R, Zivy M, Balliau T, Le Floch G, Pensec F. Deciphering the Infectious Process of Colletotrichum lupini in Lupin through Transcriptomic and Proteomic Analysis. Microorganisms. 2020; 8(10):1621. https://doi.org/10.3390/microorganisms8101621
Chicago/Turabian StyleDubrulle, Guillaume, Adeline Picot, Stéphanie Madec, Erwan Corre, Audrey Pawtowski, Riccardo Baroncelli, Michel Zivy, Thierry Balliau, Gaétan Le Floch, and Flora Pensec. 2020. "Deciphering the Infectious Process of Colletotrichum lupini in Lupin through Transcriptomic and Proteomic Analysis" Microorganisms 8, no. 10: 1621. https://doi.org/10.3390/microorganisms8101621