
Sedimentary Ancient DNA (sedaDNA) Reveals Fungal Diversity and Environmental Drivers of Community Changes throughout the Holocene in the Present Boreal Lake Lielais Svētiņu (Eastern Latvia)


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling and Chronology of the Sediment Cores
2.2. Molecular Analysis
2.3. Bioinformatics Analysis
2.4. Assigning Ecological Roles of Fungi
2.5. Statistical Analysis
2.5.1. Richness Changes between Time Periods
2.5.2. Community Composition and Ecology of Fungi
2.5.3. Past Environmental Drivers
3. Results
3.1. Fungal Sequences
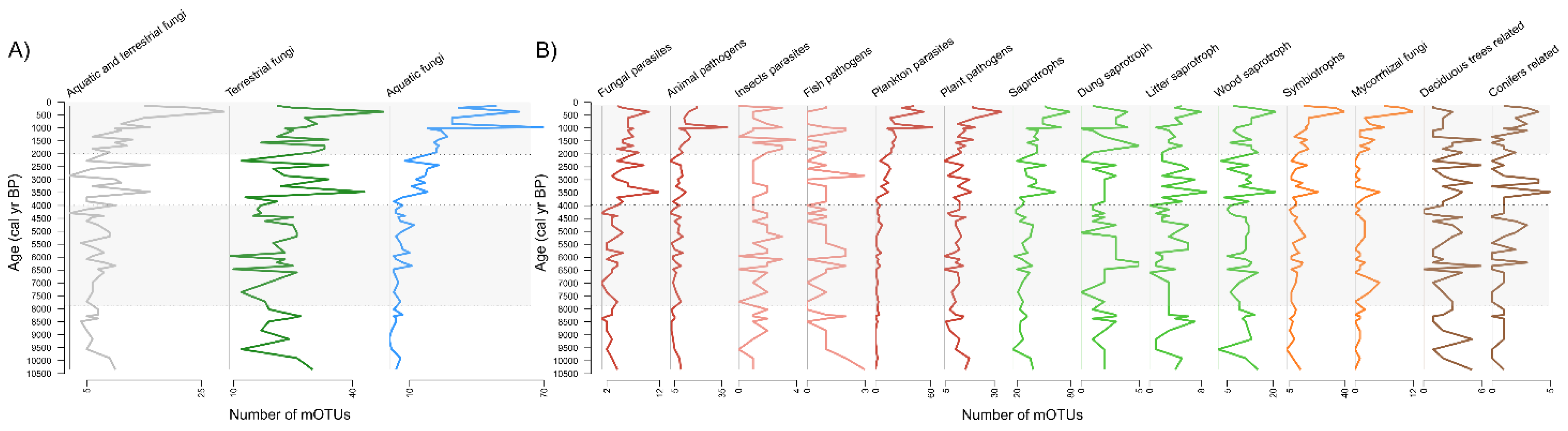
3.2. Overall Phylogenetic and Ecophysiological Richness of Fungi
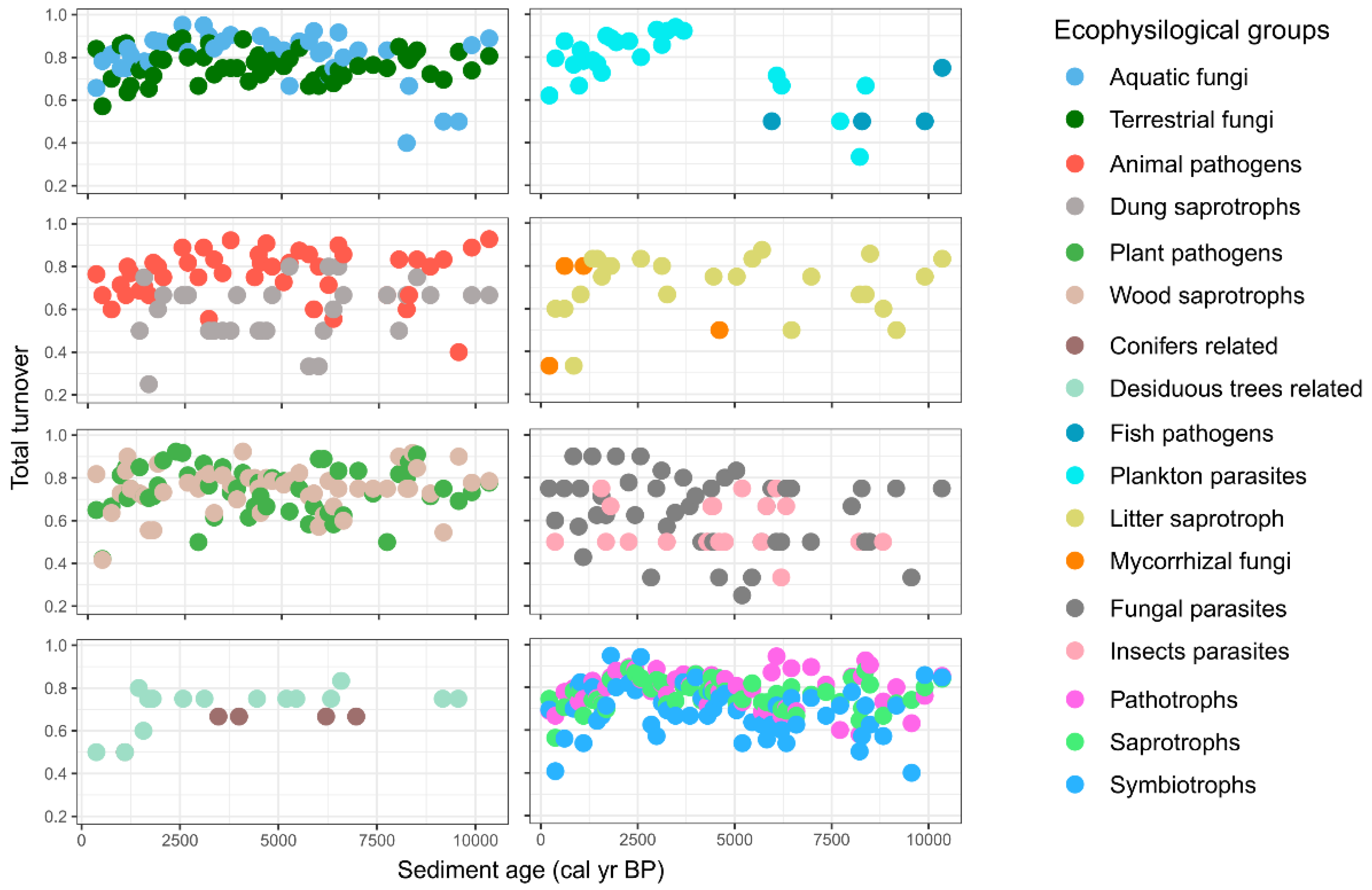
3.3. Dynamics and Development of the Fungal Community
3.4. Past Environmental Drivers
4. Discussion
4.1. Fungal Community Composition and Their Ecological Diversity
4.2. Fungal Community Changes and Paleoecological Drivers
4.3. Increased Human Impact and Change in Richness of Fungi
4.4. Challenges with Using Fungal Richness and Diversity as a Proxy for Total Paleo-Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Study Site Description
Appendix A.2. Subsampling
Appendix A.3. DNA Extraction and PCR
Appendix A.4. Bioinformatic Analysis
Appendix A.5. Statistical Analyses: GAM Model
Appendix A.6. Reproducibility of Amplicon-Based Community Descriptors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Count of mOTUs | ||||||
| Adonis (formula = binary data ~ Tech_repl + Bio_repl, data = metadata 3, permutations = 99, method = “jaccard”) | ||||||
| -Source of variation | Df | SumsOfSqs | MeanSqs | F.Model | R2 | Pr (>F) |
| Tech_repl 1 | 5 | 1.6445 | 0.32890 | 0.81151 | 0.07500 | 1.00 |
| Bio_repl 2 | 2 | 2.0450 | 1.02252 | 2.52291 | 0.09326 | 0.01 ** |
| Residuals | 45 | 18.2382 | 0.40529 | - | 0.83174 | - |
| Total | 52 | 21.9278 | - | - | 1.00000 | - |
| Raw Read Counts Data | ||||||
| Adonis (formula = read data ~ Tech_repl + Bio_repl, data = metadata 3, permutations = 99, method = “bray”) | ||||||
| -Source of variation | Df | SumsOfSqs | MeanSqs | F.Model | R2 | Pr (>F) |
| Tech_repl 1 | 5 | 1.3118 | 0.26237 | 0.6743 | 0.05793 | 1.00 |
| Bio_repl 2 | 2 | 3.8255 | 1.91274 | 4.9158 | 0.16892 | 0.01 ** |
| Residuals | 45 | 17.5095 | 0.38910 | - | 0.77315 | - |
| Total | 52 | 22.6468 | - | - | 1.00000 | - |
| (A) Count of mOTUs | m1rs = lme(chao ~ Biol 1 + Depth, random = ~1| Tech_repl 2, data = d, method = “ML”) m1r_s = lme(chao ~ Biol * Depth, random = ~1|Tech_repl, data = d, method = “ML”) anova (m1rs, m1r_s) | |||||||
|---|---|---|---|---|---|---|---|---|
| Model | df | AIC | BIC | logLik | Test | L.Ratio | p-Value | |
| Biol + Depth | m1rs | 1 | 7 | 1757.704 | 1780.897 | −871.8521 | - | - |
| Biol * Depth | m1r_s | 2 | 10 | 1742.946 | 1776.078 | −861.4731 | 1 vs. 2 20.75801 | 0.0001 |
| summary (m1r_s) | ||||||||
| Random effects: | Formula: ~1 | Tech_repl | |||||||
| (Intercept) | Residual | |||||||
| StdDev: | 4.035052 | 16.69432 | ||||||
| Fixed effects: | chao ~ Repl * Depth2 | |||||||
| Value | Std.Error | DF | t-Value | p-Value | ||||
| (Intercept) | 81.72375 | 8.33564 | 190 | 9.804133 | 0.0000 | |||
| Repl2 | −62.74254 | 11.09800 | 190 | −5.65350 | 0.0000 | |||
| Repl3 | −23.79641 | 11.14714 | 190 | −2.134754 | 0.0341 | |||
| Depth2 | −0.06593 | 0.00994 | 190 | −6.630670 | 0.0000 | |||
| Repl2:Depth2 | 0.05890 | 0.01371 | 190 | 4.295839 | 0.0000 | |||
| Repl3:Depth2 | 0.01239 | 0.01380 | 190 | 0.897395 | 0.3706 | |||
| (B) Raw Read Counts Data | m3rs = lme(read_sum ~ Biol 1 + Depth, random = ~1|Tech_repl2, data = d2, method = “ML”) m3r_s = lme(read_sum ~ Biol * Depth, random = ~1|Tech_repl, data = d2, method = “ML”) anova (m3rs, m3r_s) | |||||||
| Model | df | AIC | BIC | logLik | Test | L.Ratio | p-Value | |
| Biol + Depth | m3rs | 1 | 7 | 5253.577 | 5276.769 | −2619.789 | ||
| Biol * Depth | m3r_s | 2 | 10 | 5253.314 | 5286.446 | −2616.657 | 1 vs. 2 6.263113 | 0.0995 |
| summary(m3r_s) | ||||||||
| Random effects: | Formula: ~1 | Tech_repl | |||||||
| - | (Intercept) | Residual | - | |||||
| StdDev: | 5.547906 | 95,893.84 | ||||||
| Fixed effects: | read_sum ~ Repl * Depth2 | |||||||
| - | Value | Std.Error | DF | t-Value | p-Value | - | ||
| (Intercept) | 108,192.17 | 45,577.48 | 190 | 2.3738079 | 0.0186 | - | ||
| Repl2 | −69,843.42 | 63,663.73 | 190 | −1.0970676 | 0.2740 | - | ||
| Repl3 | 96,198.08 | 63,972.61 | 190 | 1.5037386 | 0.1343 | - | ||
| Depth2 | −71.34 | 57.09 | 190 | −1.2496175 | 0.2130 | - | ||
| Repl2:Depth2 | 84.48 | 78.64 | 190 | 1.0742489 | 0.2841 | - | ||
| Repl3:Depth2 | −105.63 | 79.22 | 190 | −1.3333252 | 0.1840 | - | ||
| -Count | Biological Replicates | Estimate | Std. Error | z Value | Pr (>|z|) |
|---|---|---|---|---|---|
| mOTUs | 2–1 | −17.684 | 3.445 | −5.133 | <1 × 10−4 *** |
| 3–1 | −15.230 | 3.471 | −4.388 | <1 × 10−4 *** | |
| 3–2 | 2.454 | 3.432 | 0.715 | 0.881 | |
| Raw Read | 2–1 | −4536 | 17,140 | −0.265 | 0.993 |
| 3–1 | 11,782 | 17,266 | 0.682 | 0.895 | |
| 3–2 | 16,317 | 17,073 | 0.956 | 0.757 |

Appendix A.7. Ecology of Fungi
Appendix A.8. Methodological Considerations
References
- Ficetola, G.F.; Poulenard, J.; Sabatier, P.; Messager, E.; Gielly, L.; Leloup, A.; Etienne, D.; Bakke, J.; Malet, E.; Fanget, B.; et al. DNA from Lake Sediments Reveals Long-Term Ecosystem Changes after a Biological Invasion. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson-Chorney, H.T.; Davis, C.S.; Poesch, M.S.; Vinebrooke, R.D.; Carli, C.M.; Taylor, M.K. Environmental DNA in lake sediment reveals biogeography of native genetic diversity. Front. Ecol. Environ. 2019, 17, 313–318. [Google Scholar] [CrossRef]
- Clarke, C.L.; Edwards, M.E.; Brown, A.G.; Gielly, L.; Lammers, Y.; Heintzman, P.D.; Ancin-Murguzur, F.J.; Bråthen, K.A.; Goslar, T.; Alsos, I.G. Holocene floristic diversity and richness in northeast Norway revealed by sedimentary ancient DNA (sedaDNA) and pollen. Boreas 2019, 48, 299–316. [Google Scholar] [CrossRef] [Green Version]
- Alsos, I.G.; Sjögren, P.; Edwards, M.E.; Landvik, J.Y.; Gielly, L.; Forwick, M.; Coissac, E.; Brown, A.G.; Jakobsen, L.V.; Føreid, M.K.; et al. Sedimentary ancient DNA from Lake Skartjørna, Svalbard: Assessing the resilience of arctic flora to Holocene climate change. Holocene 2015, 26, 627–642. [Google Scholar] [CrossRef] [Green Version]
- Lydolph, M.C.; Jacobsen, J.; Arctander, P.; Gilbert, M.T.P.; Gilichinsky, D.A.; Hansen, A.J.; Willerslev, E.; Lange, L. Beringian Paleoecology inferred from permafrost-preserved fungal DNA. Appl. Environ. Microbiol. 2005, 71, 1012–1017. [Google Scholar] [CrossRef] [Green Version]
- Bellemain, E.; Davey, M.L.; Kauserud, H.; Epp, L.S.; Boessenkool, S.; Coissac, E.; Geml, J.; Edwards, M.; Willerslev, E.; Gussarova, G.; et al. Fungal palaeodiversity revealed using high-throughput metabarcoding of ancient DNA from arctic permafrost. Environ. Microbiol. 2013, 15, 1176–1189. [Google Scholar] [CrossRef]
- Doyen, E.; Etienne, D. Ecological and human land-use indicator value of fungal spore morphotypes and assemblages. Veg. Hist. Archaeob. 2017, 26, 357–367. [Google Scholar] [CrossRef]
- Stivrins, N.; Aakala, T.; Ilvonen, L.; Pasanen, L.; Kuuluvainen, T.; Vasander, H.; Gałka, M.; Disbrey, H.R.; Liepins, J.; Holmström, L.; et al. Integrating fire-scar, charcoal and fungal spore data to study fire events in the boreal forest of northern Europe. Holocene 2019, 29, 1480–1490. [Google Scholar] [CrossRef]
- Kochkina, G.; Ivanushkina, N.; Ozerskaya, S.; Chigineva, N.; Vasilenko, O.; Firsov, S.; Spirina, E.; Gilichinsky, D. Ancient fungi in Antarctic permafrost environments. FEMS Microbiol. Ecol. 2012, 82, 501–509. [Google Scholar] [CrossRef] [Green Version]
- De Schepper, S.; Ray, J.L.; Skaar, K.S.; Sadatzki, H.; Ijaz, U.Z.; Stein, R.; Larsen, A. The potential of sedimentary ancient DNA for reconstructing past sea ice evolution. ISME J. 2019, 13, 2566–2577. [Google Scholar] [CrossRef] [Green Version]
- Kisand, V.; Talas, L.; Kisand, A.; Stivrins, N.; Reitalu, T.; Alliksaar, T.; Vassiljev, J.; Liiv, M.; Heinsalu, A.; Seppä, H.; et al. From microbial eukaryotes to metazoan vertebrates: Wide spectrum paleo-diversity in sedimentary ancient DNA over the last ~14,500 years. Geobiology 2018, 16, 628–639. [Google Scholar] [CrossRef]
- Capo, E.; Debroas, D.; Arnaud, F.; Guillemot, T.; Bichet, V.; Millet, L.; Gauthier, E.; Massa, C.; Develle, A.L.; Pignol, C.; et al. Long-term dynamics in microbial eukaryotes communities: A palaeolimnological view based on sedimentary DNA. Mol. Ecol. 2016, 25, 5925–5943. [Google Scholar] [CrossRef]
- Capo, E.; Ninnes, S.; Domaizon, I.; Bertilsson, S.; Bigler, C.; Wang, X.R.; Bindler, R.; Rydberg, J. Landscape Setting Drives the Microbial Eukaryotic Community Structure in Four Swedish Mountain Lakes over the Holocene. Microorganisms 2021, 9, 355. [Google Scholar] [CrossRef]
- Epure, L.; Meleg, I.N.; Munteanu, C.-M.; Dumitru, R.; Oana, R.; Moldovan, T. Bacterial and Fungal Diversity of Quaternary Cave Sediment Deposits. Geomicrobiol. J. 2014, 31, 116–127. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: An in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Ruiz, L.V.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A.; et al. Global diversity and geography of soil fungi. Science 2014, 346, 1256688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khomich, M.; Davey, M.L.; Kauserud, H.; Rasconi, S.; Andersen, T. Fungal communities in Scandinavian lakes along a longitudinal gradient. Fungal Ecol. 2017, 27, 36–46. [Google Scholar] [CrossRef]
- Xu, W.; Gao, Y.; Gong, L.; Li, M.; Pang, K.L.; Luo, Z.H. Fungal diversity in the deep-sea hadal sediments of the Yap Trench by cultivation and high throughput sequencing methods based on ITS rRNA gene. Deep Sea Res. Part I Oceanogr. Res. Pap. 2019, 145, 125–136. [Google Scholar] [CrossRef]
- Kagami, M.; Miki, T.; Takimoto, G. Mycoloop: Chytrids in aquatic food webs. Front. Microbiol. 2014, 5, PMC4001071. [Google Scholar] [CrossRef]
- Stivrins, N.; Kołaczek, P.; Reitalu, T.; Seppä, H.; Veski, S. Phytoplankton response to the environmental and climatic variability in a temperate lake over the last 14,500 years in eastern Latvia. J. Paleolimnol. 2015, 103–119. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, e2584. [Google Scholar] [CrossRef] [PubMed]
- Frøslev, T.G.; Kjøller, R.; Bruun, H.H.; Ejrnæs, R.; Brunbjerg, A.K.; Pietroni, C.; Hansen, A.J. Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nat. Commun. 2017, 8, 1188. [Google Scholar] [CrossRef] [PubMed]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Põlme, S.; Bahram, M.; Jacquemyn, H.; Kennedy, P.; Kohout, P.; Moora, M.; Oja, J.; Öpik, M.; Pecoraro, L.; Tedersoo, L. Host preference and network properties in biotrophic plant–fungal associations. New Phytol. 2018, 217, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Ellis, W. Plant Parasites of Europe. Available online: https://bladmineerders.nl/ (accessed on 10 September 2019).
- Cannon, P.; Kirk, P. Fungal Families of the World; CABI: Wallingford, UK, 2007; pp. 1–456. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, 2018. Available online: http://www.r-project.org/ (accessed on 29 March 2021).
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. Vegan: Community Ecology Package. R package version 2.5–4. 2019. Available online: https://cran.r-project.org/package=vegan (accessed on 29 March 2021).
- Hallett, L.M.; Jones, S.K.; MacDonald, A.A.M.; Jones, M.B.; Flynn, D.F.B.; Ripplinger, J.; Slaughter, P.; Gries, C.; Collins, S.L. Codyn: An R package of community dynamics metrics. Methods Ecol. Evol. 2016, 7, 1146–1151. [Google Scholar] [CrossRef]
- Collins, S.L.; Micheli, F.; Hartt, L.; Collins, S.L. A Method to Determine Rates and Patterns of Variability in Ecological Communities. Oikos 2000, 91, 285–293. Available online: https://www.jstor.org/stable/3547549 (accessed on 29 March 2021). [CrossRef]
- Gross, K.; Cardinale, B.J.; Fox, J.W.; Gonzalez, A.; Loreau, M.; Polley, H.W.; Reich, P.B.; Van Ruijven, J. Species richness and the temporal stability of biomass production: A new analysis of recent biodiversity experiments. Am. Nat. 2014, 183, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Paulson, J.N.; Colin Stine, O.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stivrins, N.; Kalnina, L.; Veski, S.; Zeimule, S. Local and regional Holocene vegetation dynamics at two sites in eastern Latvia. Boreal Environ. Res. 2014, 19, 310–322. [Google Scholar]
- Kagami, M.; De Bruin, A.; Ibelings, B.W.; Van Donk, E. Parasitic chytrids: Their effects on phytoplankton communities and food-web dynamics. Hydrobiologia 2007, 578, 113–129. [Google Scholar] [CrossRef] [Green Version]
- Větrovský, T.; Kohout, P.; Kopecký, M.; Machac, A.; Man, M.; Bahnmann, B.D.; Brabcová, V.; Choi, J.; Meszárošová, L.; Human, Z.R.; et al. A meta-analysis of global fungal distribution reveals climate-driven patterns. Nat. Commun. 2019, 10, 5142. [Google Scholar] [CrossRef] [Green Version]
- Feurdean, A.; Veski, S.; Florescu, G.; Vannière, B.; Pfeiffer, M.; O’Hara, R.B.; Stivrins, N.; Amon, L.; Heinsalu, A.; Vassiljev, J.; et al. Broadleaf deciduous forest counterbalanced the direct effect of climate on Holocene fire regime in hemiboreal/boreal region (NE Europe). Quat. Sci. Rev. 2017, 169, 378–390. [Google Scholar] [CrossRef]
- Röhl, O.; Peršoh, D.; Mittelbach, M.; Elbrecht, V.; Brachmann, A.; Nuy, J.; Boenigk, J.; Leese, F.; Begerow, D. Distinct sensitivity of fungal freshwater guilds to water quality. Mycol. Prog. 2017, 16, 155–169. [Google Scholar] [CrossRef]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Drenkhan, R.; Pritsch, K.; Buegger, F.; Padari, A.; Hagh-Doust, N.; Mikryukov, V.; Gohar, D.; et al. Regional-Scale In-Depth Analysis of Soil Fungal Diversity Reveals Strong pH and Plant Species Effects in Northern Europe. Front. Microbiol. 2020, 11, 1953. [Google Scholar] [CrossRef]
- Giguet-Covex, C.; Ficetola, G.F.; Walsh, K.; Poulenard, J.; Bajard, M.; Fouinat, L.; Sabatier, P.; Gielly, L.; Messager, E.; Develle, A.L.; et al. New insights on lake sediment DNA from the catchment: Importance of taphonomic and analytical issues on the record quality. Sci. Rep. 2019, 9, 14676. [Google Scholar] [CrossRef]
- Wurzbacher, C.; Rösel, S.; Rychła, A.; Grossart, H.P. Importance of Saprotrophic Freshwater Fungi for Pollen Degradation. PLoS ONE 2014, 9, e94643. [Google Scholar] [CrossRef] [Green Version]
- Grossart, H.P.; Wurzbacher, C.; James, T.Y.; Kagami, M. Discovery of dark matter fungi in aquatic ecosystems demands a reappraisal of the phylogeny and ecology of zoosporic fungi. Fungal Ecol. 2016, 19, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Gleason, F.H.; Kagami, M.; Lefevre, E.; Sime-Ngando, T. The ecology of chytrids in aquatic ecosystems: Roles in food web dynamics. Fungal Biol. Rev. 2008, 22, 17–25. [Google Scholar] [CrossRef]
- Wurzbacher, C.; Warthmann, N.; Bourne, E.C.; Attermeyer, K.; Allgaier, M.; Powell, J.R.; Detering, H.; Mbedi, S.; Grossart, H.P.; Monaghan, M.T. High habitat-specificity in fungal communities in oligo-mesotrophic, temperate Lake Stechlin (North-East Germany). MycoKeys 2016, 16, 17–44. [Google Scholar] [CrossRef] [Green Version]
- Ishida, S.; Nozaki, D.; Grossart, H.P.; Kagami, M. Novel basal, fungal lineages from freshwater phytoplankton and lake samples. Environ. Microbiol. Rep. 2015, 7, 435–441. [Google Scholar] [CrossRef]
- Jones, M.D.M.; Forn, I.; Gadelha, C.; Egan, M.J.; Bass, D.; Massana, R.; Richards, T.A. Discovery of novel intermediate forms redefines the fungal tree of life. Nature 2011, 474, 200–204. [Google Scholar] [CrossRef]
- Aakala, T. Forest fire histories and tree age structures in Värriö and Maltio Strict Nature Reserves, northern Finland. Boreal Environ. Res. 2018, 23, 209–219. [Google Scholar]
- Hurt, R.A.; Qiu, X.; Wu, L.; Roh, Y.; Palumbo, A.V.; Tiedje, J.M.; Zhou, J. Simultaneous Recovery of RNA and DNA from Soils and Sediments. Appl. Environ. Microbiol. 2001, 67, 4495–4503. [Google Scholar] [CrossRef] [Green Version]
- Lakay, F.M.; Botha, A.; Prior, B.A. Comparative analysis of environmental DNA extraction and purification methods from different humic acid-rich soils. J. Appl. Microbiol. 2007, 102, 265–273. [Google Scholar] [CrossRef]
- Stadler, M.; Lambert, C.; Wibberg, D.; Kalinowski, J.; Cox, R.J.; Kolařík, M.; Kuhnert, E. Intragenomic polymorphisms in the ITS region of high-quality genomes of the Hypoxylaceae (Xylariales, Ascomycota). Mycol. Progress 2020, 19, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Seglins, V.; Kalnina, L.; Lacis, A. The Lubans Plain, Latvia, as a reference area for long term studies of human impact on the environment. PACT 1999, 57, 105–129. [Google Scholar]
- Zelčs, V.; Markots, A. Deglaciation history of Latvia. In Quaternary Glaciations—Extent and Chronology of Glaciations, 1st ed.; Ehlers, J., Gibbard, P.L., Eds.; Elsiever: Amsterdam, The Netherlands, 2004; Volume 2, pp. 225–243. [Google Scholar]
- Veski, S.; Amon, L.; Heinsalu, A.; Reitalu, T.; Saarse, L.; Stivrins, N.; Vassiljev, J. Lateglacial vegetation dynamics in the eastern Baltic region between 14,500 and 11 400 cal yr BP: A complete record since the Bølling (GI-1e) to the Holocene. Quat. Sci. Rev. 2012, 40, 39–53. [Google Scholar] [CrossRef]
- Unterseher, M.; Jumpponen, A.; Öpik, M.; Tedersoo, L.; Moora, M.; Dormann, C.F.; Schnittler, M. Species abundance distributions and richness estimations in fungal metagenomics—Lessons learned from community ecology. Mol. Ecol. 2011, 20, 275–285. [Google Scholar] [CrossRef]
- Pinheiro, J.; Bates, D.; DebRoy, S.; Sarkar, D.; R Core Team. Nlme: Linear and nonlinear mixed effects models. R package version 3. 2019, pp. 1–140. Available online: https://CRAN.R-project.org/package=nlme (accessed on 15 October 2019).
- Hothorn, T.; Bretz, F.; Westfall, P.; Heiberger, R.M.; Schuetzenmeister, A.; Scheibe, S. Multcomp: Simultaneous inference in general parametric models. R package version 1.4–5. 2016. Available online: http://cran.stat.sfu.ca/web/packages/multcomp/multcomp.pdf (accessed on 29 March 2021).
- Frenken, T.; Alacid, E.; Berger, S.A.; Bourne, E.C.; Gerphagnon, M.; Grossart, H.-P.; Gsell, A.S.; Ibelings, B.W.; Kagami, M.; Küpper, F.C. Integrating chytrid fungal parasites into plankton ecology: Research gaps and needs. Environ. Microbiol. 2017, 19, 3802–3822. [Google Scholar] [CrossRef] [Green Version]






| Ecological Group | Rate Change 1 | Synchrony 2 | Variance Ratio 3 |
|---|---|---|---|
| Water environment | 0.04 | 0.249 | 16.7 |
| Terrestrial environment | 0.013 | 0.058 | 3.2 |
| Both environments | 0.017 | 0.121 | 3.4 |
| Pathotroph | 0.038 | 0.166 | 15 |
| Animal pathogen | 0.017 | 0.175 | 4.9 |
| Insects parasite | 0.006 | −0.166 | 0.5 |
| Fish pathogen | 0.003 | −0.276 | 0.3 |
| Plankton parasite | 0.045 | 0.269 | 17.8 |
| Fungal parasite | 0.009 | 0.062 | 1.7 |
| Plant pathogen | 0.015 | 0.077 | 2.7 |
| Saprotroph | 0.025 | 0.09 | 6.3 |
| Litter saprotroph | 0.004 | 0.03 | 1.1 |
| Wood saprotroph | 0.009 | 0.046 | 1.8 |
| Dung saprotroph | 0.004 | −0.037 | 0.7 |
| Symbiotroph | 0.018 | 0.134 | 5.2 |
| Lichen symbiont | 0.0007 | −0.326 | 0.1 |
| Mycorrhizal fungi | 0.047 | 0.141 | 23.7 |
| Conifer-related fungi | 0.001 | −0.028 | 0.7 |
| Deciduous tree-related fungi | 0.0002 | 0.002 | 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talas, L.; Stivrins, N.; Veski, S.; Tedersoo, L.; Kisand, V. Sedimentary Ancient DNA (sedaDNA) Reveals Fungal Diversity and Environmental Drivers of Community Changes throughout the Holocene in the Present Boreal Lake Lielais Svētiņu (Eastern Latvia). Microorganisms 2021, 9, 719. https://doi.org/10.3390/microorganisms9040719
Talas L, Stivrins N, Veski S, Tedersoo L, Kisand V. Sedimentary Ancient DNA (sedaDNA) Reveals Fungal Diversity and Environmental Drivers of Community Changes throughout the Holocene in the Present Boreal Lake Lielais Svētiņu (Eastern Latvia). Microorganisms. 2021; 9(4):719. https://doi.org/10.3390/microorganisms9040719
Chicago/Turabian StyleTalas, Liisi, Normunds Stivrins, Siim Veski, Leho Tedersoo, and Veljo Kisand. 2021. "Sedimentary Ancient DNA (sedaDNA) Reveals Fungal Diversity and Environmental Drivers of Community Changes throughout the Holocene in the Present Boreal Lake Lielais Svētiņu (Eastern Latvia)" Microorganisms 9, no. 4: 719. https://doi.org/10.3390/microorganisms9040719