
Diversity of Unusual Ribosomal Genes and Ecological Origin of Rice (Oryza spp.)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials Used in the Study
2.2. DNA and RNA Extraction
2.3. Gene Sequencing
2.4. Probes (RC and RDF) Labeled and Northern Hybridization
2.5. Terminated Restriction Fragment Length Polymorphism (T-RFLP)
2.6. Sequences Data Analysis
3. Results
3.1. Phylogeny Inferred from Concatenated Sequences of the Genus Oryza
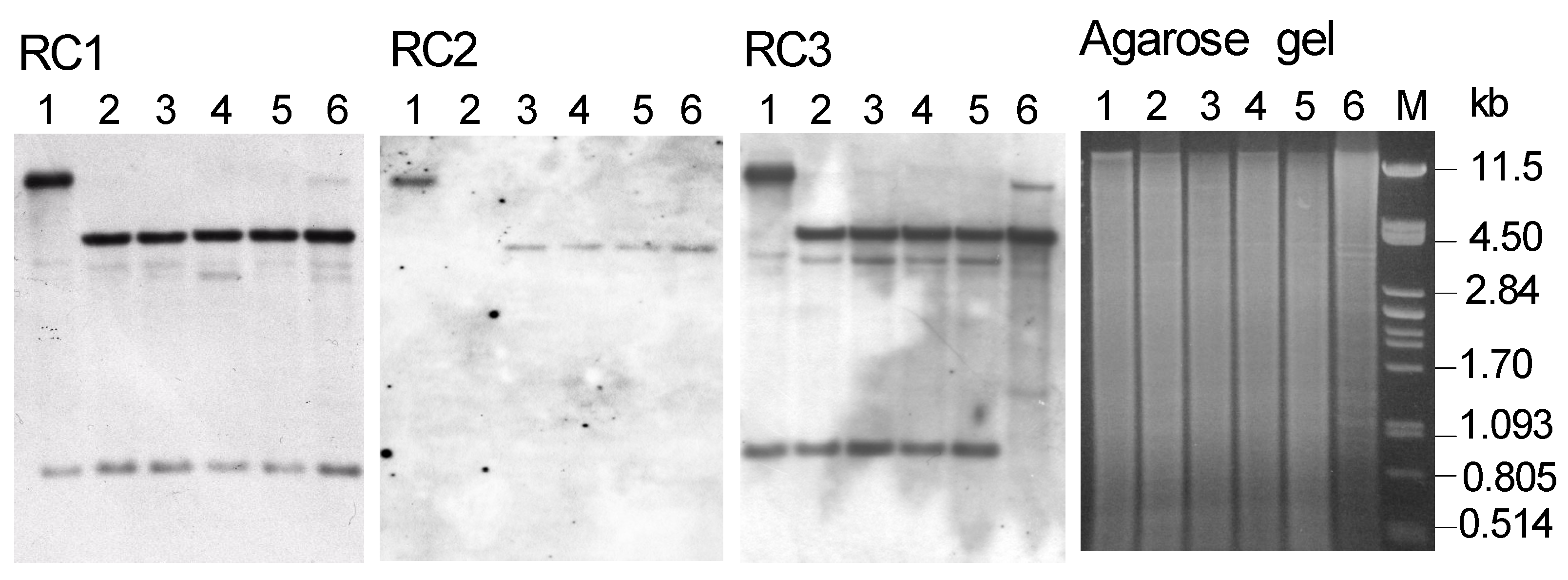
3.2. Validation of the PCR Sequence Artifacts through Southern Hybridization
3.3. Fragments Hybridization of RDF1-4
3.4. Analyses of 28S Genes in Ribosome of O. officinalis
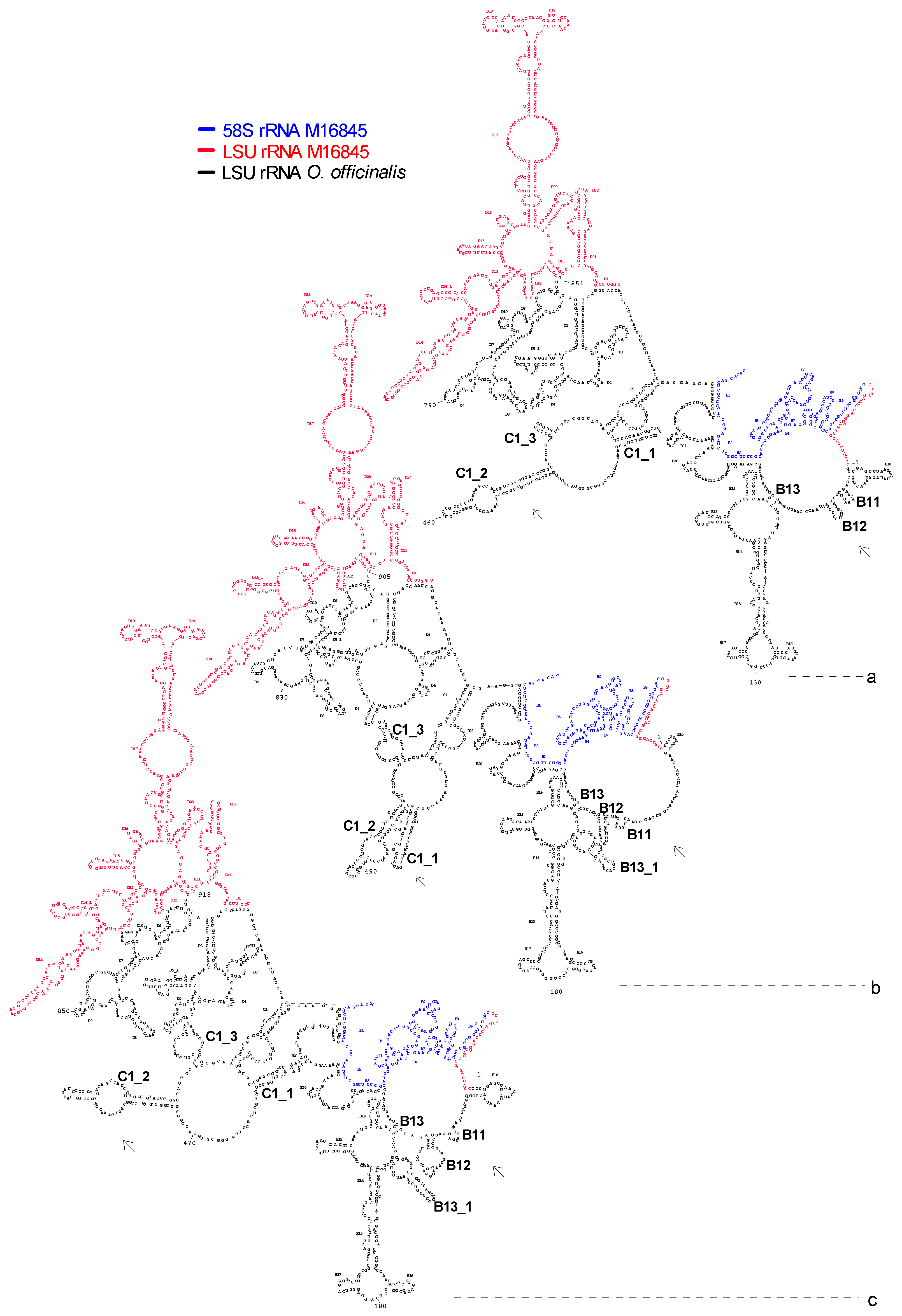
3.5. Conservation of RNA Secondary Structure
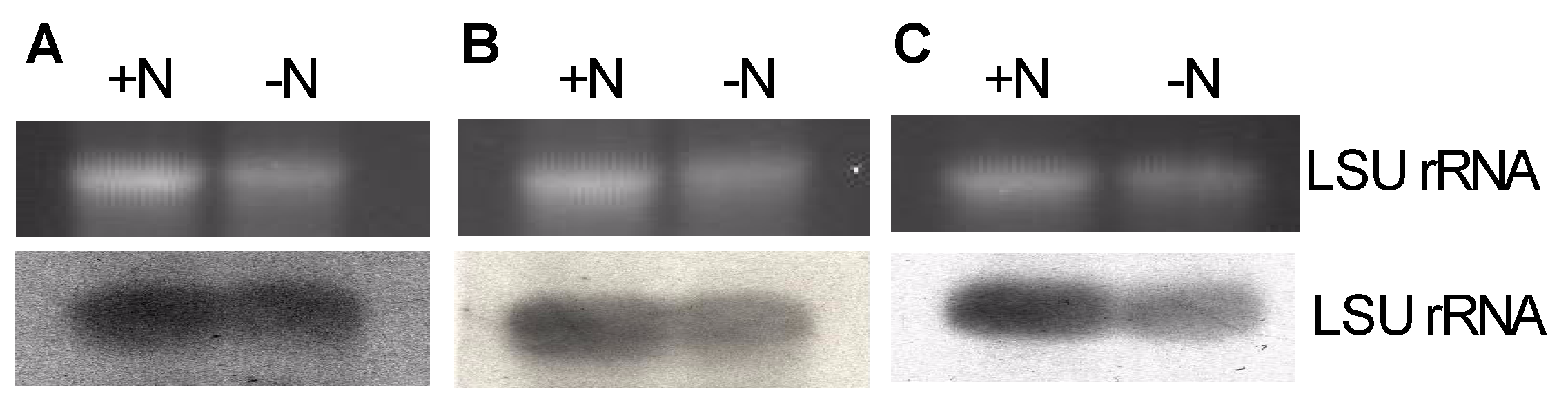
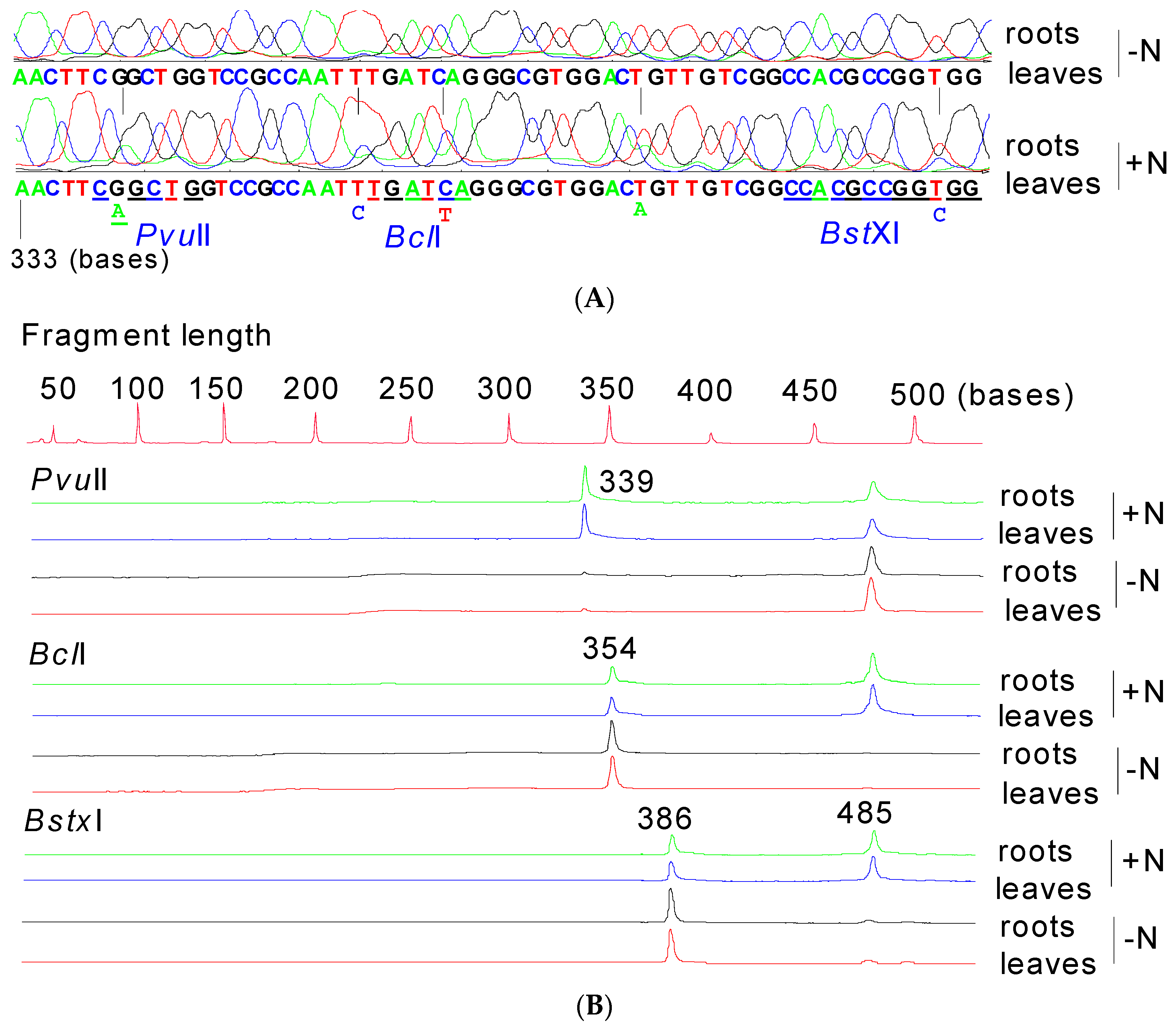
3.6. Differential Expression of 28S Ribosomal Genes with and without Fertilization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wing, R.A.; Purugganan, M.D.; Zhang, Q. The rice genome revolution: From an ancient grain to Green Super Rice. Nat. Rev. Genet. 2018, 19, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Stein, J.C.; Yu, Y.; Copetti, D.; Zwickl, D.J.; Zhang, L.; Zhang, C.; Chougule, K.; Gao, D.; Iwata, A.; Goicoechea, J.L. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018, 50, 285–296. [Google Scholar] [CrossRef]
- Tabassum, J.; Raza, Q.; Riaz, A.; Ahmad, S.; Rashid, M.A.R.; Javed, M.A.; Ali, Z.; Kang, F.; Khan, I.A.; Atif, R.M. Exploration of the genomic atlas of Dof transcription factor family across genus Oryza provides novel insights on rice breeding in changing climate. Front. Plant Sci. 2022, 13, 1004359. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.L.; Zhao, Z. Archaeological and genetic insights into the origins of domesticated rice. Proc. Natl. Acad. Sci. USA 2014, 111, 6190–6197. [Google Scholar] [CrossRef]
- Zhang, T.; Li, X.; Zhao, Z.; Wu, R.; Yang, Z.; He, G. Sequencing and Genomic Analysis of Sorghum DNA Introgression Variant Line R21 and Recipient Rice Jin Hui 1 Revealed Repetitive Element Variation. Int. J. Mol. Sci. 2022, 23, 11864. [Google Scholar] [CrossRef]
- Konikkat, S.; Woolford, J.L., Jr. Principles of 60S ribosomal subunit assembly emerging from recent studies in yeast. Biochem. J. 2017, 474, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Mitterer, V.; Pertschy, B. RNA folding and functions of RNA helicases in ribosome biogenesis. RNA Biol. 2022, 19, 781–810. [Google Scholar] [CrossRef]
- Espinar-Marchena, F.J.; Fernández-Fernández, J.; Rodríguez-Galán, O.; Fernández-Pevida, A.; Babiano, R.; de la Cruz, J. Role of the yeast ribosomal protein L16 in ribosome biogenesis. FEBS J. 2016, 283, 2968–2985. [Google Scholar] [CrossRef]
- Wan, K.; Yabuki, Y.; Mizuta, K. Roles of Ebp2 and ribosomal protein L36 in ribosome biogenesis in Saccharomyces cerevisiae. Curr. Genet. 2015, 61, 31–41. [Google Scholar] [CrossRef]
- Wang, X.; Yue, Z.; Xu, F.; Wang, S.; Hu, X.; Dai, J.; Zhao, G. Coevolution of ribosomal RNA expansion segment 7L and assembly factor Noc2p specializes the ribosome biogenesis pathway between Saccharomyces cerevisiae and Candida albicans. Nucleic Acids Res. 2021, 49, 4655–4667. [Google Scholar] [CrossRef]
- Gόmez Ramos, L.M.; Degtyareva, N.N.; Kovacs, N.A.; Holguin, S.Y.; Jiang, L.; Petrov, A.S.; Biesiada, M.; Hu, M.Y.; Purzycka, K.J.; Arya, D.P. Eukaryotic ribosomal expansion segments as antimicrobial targets. Biochemistry 2017, 56, 5288–5299. [Google Scholar] [CrossRef]
- Leppek, K.; Fujii, K.; Quade, N.; Susanto, T.T.; Boehringer, D.; Lenarčič, T.; Xue, S.; Genuth, N.R.; Ban, N.; Barna, M. Gene-and species-specific Hox mRNA translation by ribosome expansion segments. Mol. Cell 2020, 80, 980–995. [Google Scholar] [CrossRef]
- Paul, B.; Raj, K.K.; Murali, T.S.; Satyamoorthy, K. Species-specific genomic sequences for classification of bacteria. Comput. Biol. Med. 2020, 123, 103874. [Google Scholar] [CrossRef]
- Raza, Q.; Rashid, M.A.R.; Waqas, M.; Ali, Z.; Rana, I.A.; Khan, S.H.; Khan, I.A.; Atif, R.M. Genomic diversity of aquaporins across genus Oryza provides a rich genetic resource for development of climate resilient rice cultivars. BMC Plant Biol. 2023, 23, 172. [Google Scholar] [CrossRef]
- Chen, E.; Huang, X.; Tian, Z.; Wing, R.A.; Han, B. The genomics of Oryza species provides insights into rice domestication and heterosis. Annu. Rev. Plant Biol. 2019, 70, 639–665. [Google Scholar] [CrossRef]
- Wei, C.; Wang, Z.; Wang, J.; Teng, J.; Shen, S.; Xiao, Q.; Bao, S.; Feng, Y.; Zhang, Y.; Li, Y. Conversion between 100-million-year-old duplicated genes contributes to rice subspecies divergence. BMC Genom. 2021, 22, 460. [Google Scholar] [CrossRef]
- He, L.; Hörandl, E. Does polyploidy inhibit sex chromosome evolution in angiosperms? Front. Plant Sci. 2022, 13, 976765. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.E.; Tang, H.; Burke, J.M.; Paterson, A.H. GC content of plant genes is linked to past gene duplications. PLoS ONE 2022, 17, e0261748. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, Y.; Lv, Y.; Pu, Q.; Li, J.; Zhang, Y.; Deng, X.; Wang, M.; Wang, J.; Tao, D. Interspecific Hybridization Is an Important Driving Force for Origin and Diversification of Asian Cultivated Rice Oryza sativa L. Front. Plant Sci. 2022, 13, 932737. [Google Scholar] [CrossRef]
- Palaniyappan, S.; Arunachalam, P.; Banumathy, S.; Mini, M.; Muthuramu, S. Genetic divergence and clustering studies in advanced breeding lines of rice (Oryza sativa L.). Electron. J. Plant Breed. 2020, 11, 499–504. [Google Scholar]
- Morgenstern, B.; Zhu, B.; Horwege, S.; Chris, A.L. Estimating evolutionary distances between genomic sequences from spaced-word matches. Algorithms Mol. Biol. 2015, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Han, Q.; Chen, F.; Li, M.; Balbuena, T.S.; Zhao, Y. Phylogenomics as an effective approach to untangle cross-species hybridization event: A case study in the family Nymphaeaceae. Front. Genet. 2022, 13, 1031705. [Google Scholar] [CrossRef]
- Sun, K.; Li, D.; Xia, A.; Zhao, H.; Wen, Q.; Jia, S.; Wang, J.; Yang, G.; Zhou, D.; Huang, C. Targeted Identification of Rice Grain-Associated Gene Allelic Variation through Mutation Induction, Targeted Sequencing, and Whole Genome Sequencing Combined with a Mixed-Samples Strategy. Rice 2022, 15, 57. [Google Scholar] [CrossRef]
- Zeng, H.; Chen, X.; Li, H.; Zhang, J.; Wei, Z.; Wang, Y. Interpopulation differences of retroduplication variations (RDVs) in rice retrogenes and their phenotypic correlations. Comput. Struct. Biotechnol. J. 2021, 19, 600–611. [Google Scholar] [CrossRef]
- Parks, M.M.; Kurylo, C.M.; Batchelder, J.E.; Theresa Vincent, C.; Blanchard, S.C. Implications of sequence variation on the evolution of rRNA. Chromosome Res. 2019, 27, 89–93. [Google Scholar] [CrossRef]
- Shi, C.; Li, W.; Zhang, Q.-J.; Zhang, Y.; Tong, Y.; Li, K.; Liu, Y.-L.; Gao, L.-Z. The draft genome sequence of an upland wild rice species, Oryza granulata. Sci. Data 2020, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-Z.; Liu, Y.-L.; Zhang, D.; Li, W.; Gao, J.; Liu, Y.; Li, K.; Shi, C.; Zhao, Y.; Zhao, Y.-J. Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2019, 2, 278. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesh, D.; Kumar, M.P.; Chandrakanth, R.; Devaki, N. Molecular diversity of internal transcribed spacer among the monoconidial isolates of Magnaporthe oryzae isolated from rice in Southern Karnataka, India. J. Genet. Eng. Biotechnol. 2018, 16, 631–638. [Google Scholar] [CrossRef]
- Jayaswal, P.K.; Dogra, V.; Shanker, A.; Sharma, T.R.; Singh, N.K. A tree of life based on ninety-eight expressed genes conserved across diverse eukaryotic species. PLoS ONE 2017, 12, e0184276. [Google Scholar] [CrossRef]
- Zou, X.-H.; Du, Y.-S.; Tang, L.; Xu, X.-W.; Doyle, J.J.; Sang, T.; Ge, S. Multiple origins of BBCC allopolyploid species in the rice genus (Oryza). Sci. Rep. 2015, 5, 14876. [Google Scholar] [CrossRef]
- Zhang, W.; Sun, Y.; Liu, J.; Xu, C.; Zou, X.; Chen, X.; Liu, Y.; Wu, P.; Yang, X.; Zhou, S. DNA barcoding of Oryza: Conventional, specific, and super barcodes. Plant Mol. Biol. 2021, 105, 215–228. [Google Scholar] [CrossRef]
- Jain, R.; Jenkins, J.; Shu, S.; Chern, M.; Martin, J.A.; Copetti, D.; Duong, P.Q.; Pham, N.T.; Kudrna, D.A.; Talag, J. Genome sequence of the model rice variety KitaakeX. BMC Genom. 2019, 20, 905. [Google Scholar] [CrossRef]
- Luo, S.; Peng, J.; Li, K.; Wang, M.; Kuang, H. Contrasting evolutionary patterns of the Rp1 resistance gene family in different species of Poaceae. Mol. Biol. Evol. 2011, 28, 313–325. [Google Scholar] [CrossRef]
- Piya, A.A.; DeGiorgio, M.; Assis, R. Predicting gene expression divergence between single-copy orthologs in two species. Genome Biol. Evol. 2023, 15, evad078. [Google Scholar] [CrossRef]
- Zhao, D.; Hamilton, J.P.; Hardigan, M.; Yin, D.; He, T.; Vaillancourt, B.; Reynoso, M.; Pauluzzi, G.; Funkhouser, S.; Cui, Y. Analysis of ribosome-associated mRNAs in rice reveals the importance of transcript size and GC content in translation. G3 Genes Genomes Genet. 2017, 7, 203–219. [Google Scholar] [CrossRef]
- Siwaszek, A.; Ukleja, M.; Dziembowski, A. Proteins involved in the degradation of cytoplasmic mRNA in the major eukaryotic model systems. RNA Biol. 2014, 11, 1122–1136. [Google Scholar] [CrossRef]
- Deng, H.; Cheema, J.; Zhang, H.; Woolfenden, H.; Norris, M.; Liu, Z.; Liu, Q.; Yang, X.; Yang, M.; Deng, X. Rice in vivo RNA structurome reveals RNA secondary structure conservation and divergence in plants. Mol. Plant 2018, 11, 607–622. [Google Scholar] [CrossRef]
- McGee, J.P.; Armache, J.-P.; Lindner, S.E. Ribosome heterogeneity and specialization of Plasmodium parasites. PLoS Pathog. 2023, 19, e1011267. [Google Scholar] [CrossRef] [PubMed]
- Burgarella, C.; Lorenzo, Z.; Jabbour-Zahab, R.; Lumaret, R.; Guichoux, E.; Petit, R.; Soto, A.; Gil, L. Detection of hybrids in nature: Application to oaks (Quercus suber and Q. ilex). Heredity 2009, 102, 442–452. [Google Scholar] [CrossRef] [PubMed]
- He, N.; Wu, R.; Pan, X.; Peng, L.; Sun, K.; Zou, T.; Zhu, H.; Zeng, R.; Liu, Z.; Liu, G. Development and trait evaluation of chromosome single-segment substitution lines of O. meridionalis in the background of O. sativa. Euphytica 2017, 213, 281. [Google Scholar] [CrossRef]
- Kopecký, D.; Martín, A.; Smýkal, P. Interspecific hybridization and plant breeding: From historical retrospective through work of Mendel to current crops. Czech J. Genet. Plant Breed. 2022, 58, 113–126. [Google Scholar] [CrossRef]
- Vemireddy, L.R.; Noor, S.; Satyavathi, V.; Srividhya, A.; Kaliappan, A.; Parimala, S.; Bharathi, P.M.; Deborah, D.A.; Rao, K.S.; Shobharani, N. Discovery and mapping of genomic regions governing economically important traits of Basmati rice. BMC Plant Biol. 2015, 15, 207. [Google Scholar] [CrossRef] [PubMed]
- Siddiq, E.; Vemireddy, L.; Nagaraju, J. Basmati rices: Genetics, breeding and trade. Agric. Res. 2012, 1, 25–36. [Google Scholar] [CrossRef]
- Reddy, M.M.; Ulaganathan, K. Draft genome sequence of Oryza sativa elite indica cultivar RP Bio-226. Front. Plant Sci. 2015, 6, 896. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. A draft genome, resequencing, and metabolomes reveal the genetic background and molecular basis of the nutritional and medicinal properties of loquat (Eriobotrya japonica (Thunb.) Lindl). Hortic. Res. 2021, 8, 231. [Google Scholar] [CrossRef]
- Yang, X.; Fuller, D.Q.; Huan, X.; Perry, L.; Li, Q.; Li, Z.; Zhang, J.; Ma, Z.; Zhuang, Y.; Jiang, L. Barnyard grasses were processed with rice around 10,000 years ago. Sci. Rep. 2015, 5, 16251. [Google Scholar] [CrossRef]
- Dai, S.-F.; Zhu, X.-G.; Hutang, G.-R.; Li, J.-Y.; Tian, J.-Q.; Jiang, X.-H.; Zhang, D.; Gao, L.-Z. Genome size variation and evolution driven by transposable elements in the genus oryza. Front. Plant Sci. 2022, 13, 921937. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.L. Gene family evolution in green plants with emphasis on the origination and evolution of a rabidopsis thaliana genes. Plant J. 2013, 73, 941–951. [Google Scholar] [CrossRef]
- Ohmido, N.; Fukui, K.; Kinoshita, T. Recent advances in rice genome and chromosome structure research by fluorescence in situ hybridization (FISH). Proc. Jpn. Acad. Ser. B 2010, 86, 103–116. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, L.; Columbus, J.T.; Hu, Y.; Zhao, Y.; Tang, L.; Guo, Z.; Chen, W.; McKain, M.; Bartlett, M. A well-supported nuclear phylogeny of Poaceae and implications for the evolution of C4 photosynthesis. Mol. Plant 2022, 15, 755–777. [Google Scholar] [CrossRef]
- Gaikwad, K.B.; Singh, N.; Kaur, P.; Rani, S.; Babu, H.P.; Singh, K. Deployment of wild relatives for genetic improvement in rice (Oryza sativa). Plant Breed. 2021, 140, 23–52. [Google Scholar] [CrossRef]
- Coiffard, C.; Kardjilov, N.; Manke, I.; Bernardes-de-Oliveira, M.E. Fossil evidence of core monocots in the Early Cretaceous. Nat. Plants 2019, 5, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, K.L.; Kinney, M.S.; Stuart, S.A.; Maurin, O.; Mathews, S.; Chase, M.W.; Gandolfo, M.A.; Pires, J.C. Phylogenetics, divergence times and diversification from three genomic partitions in monocots. Bot. J. Linn. Soc. 2015, 178, 375–393. [Google Scholar] [CrossRef]
- Dellaporta, S.L.; Wood, J.; Hicks, J.B. A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep. 1983, 1, 19–21. [Google Scholar] [CrossRef]
- Yang, Y.-W.; Lai, K.-N.; Tai, P.-Y.; Li, W.-H. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J. Mol. Evol. 1999, 48, 597–604. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sp. Name | IRGC Accession No. | Chromosome No. | Genomic Type |
|---|---|---|---|---|
| 1 | O. alta | ACC105143 | 48 | CCDD |
| 2 | O. australiensis | ACC100882 | 24 | EE |
| 3 | O. glaberrima | TOG5674 | 24 | AA |
| 4 | O. grandiglumis | ACC105669 | 48 | CCDD |
| 5 | O. granulata | ACC102119 | 24 | GG |
| 6 | O. latifolia | ACC100190 | 48 | CCDD |
| 7 | O. longiglumis | ACC105148 | 48 | HHJJ |
| 8 | O. longistaminata | ACC101140 | 24 | AA |
| 9 | O. malampuzhaensis | ACC105329 | 48 | BBCC |
| 10 | O. minuta | ACC101141 | 48 | BBCC |
| 11 | O. nivara | ACC105763 | 24 | AA |
| 12 | O. officinalis | ACC101399 | 24 | CC |
| 13 | O. punctata | ACC105690 | 24, 48 | BB, BBCC |
| 14 | O. rhizomatis | ACC105432 | 24 | CC |
| 15 | O. ridleyi | ACC100821 | 48 | HHJJ |
| 16 | O. indica | IR36 | 24 | AA |
| 17 | O. rufipogon | ACC106423 | 24 | AA |
| Taxon Pair a | Average Distance | Divergence Time (My Ago) b |
|---|---|---|
| Monocots (106) vs. dicots (09) | 0.2214 ± 0.0163 | 170–235 |
| RC1 (41) vs. RC3 (15) | 0.1862 ± 0.0205 | 143–198 |
| RC1 (41) vs. RC2 (14) | 0.1990 ± 0.0165 | 153–211 |
| RC2 (14) vs. RC3 (15) | 0.1785 ± 0.0174 | 137–190 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, X.; Peng, G.; Muhammad, S.; Kaleem, S.; Jan, M.; Munir, R.; Chen, X.; Khattak, A.A.; Abbas, A.A.; Chen, Y.; et al. Diversity of Unusual Ribosomal Genes and Ecological Origin of Rice (Oryza spp.). Agriculture 2024, 14, 265. https://doi.org/10.3390/agriculture14020265
Tan X, Peng G, Muhammad S, Kaleem S, Jan M, Munir R, Chen X, Khattak AA, Abbas AA, Chen Y, et al. Diversity of Unusual Ribosomal Genes and Ecological Origin of Rice (Oryza spp.). Agriculture. 2024; 14(2):265. https://doi.org/10.3390/agriculture14020265
Chicago/Turabian StyleTan, Xiyu, Guixiang Peng, Sajid Muhammad, Sidra Kaleem, Mehmood Jan, Raheel Munir, Xiaoyuan Chen, Arif Ali Khattak, Abid Ali Abbas, Yihang Chen, and et al. 2024. "Diversity of Unusual Ribosomal Genes and Ecological Origin of Rice (Oryza spp.)" Agriculture 14, no. 2: 265. https://doi.org/10.3390/agriculture14020265