
Population Genomics of Domesticated Cucurbita ficifolia Reveals a Recent Bottleneck and Low Gene Flow with Wild Relatives
 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Genome Sequencing and Assembly
2.3. GBS Procedure
2.4. Reference-Guided Read Mapping
2.5. Data Analysis
3. Results
3.1. Cucurbita ficifolia Genome
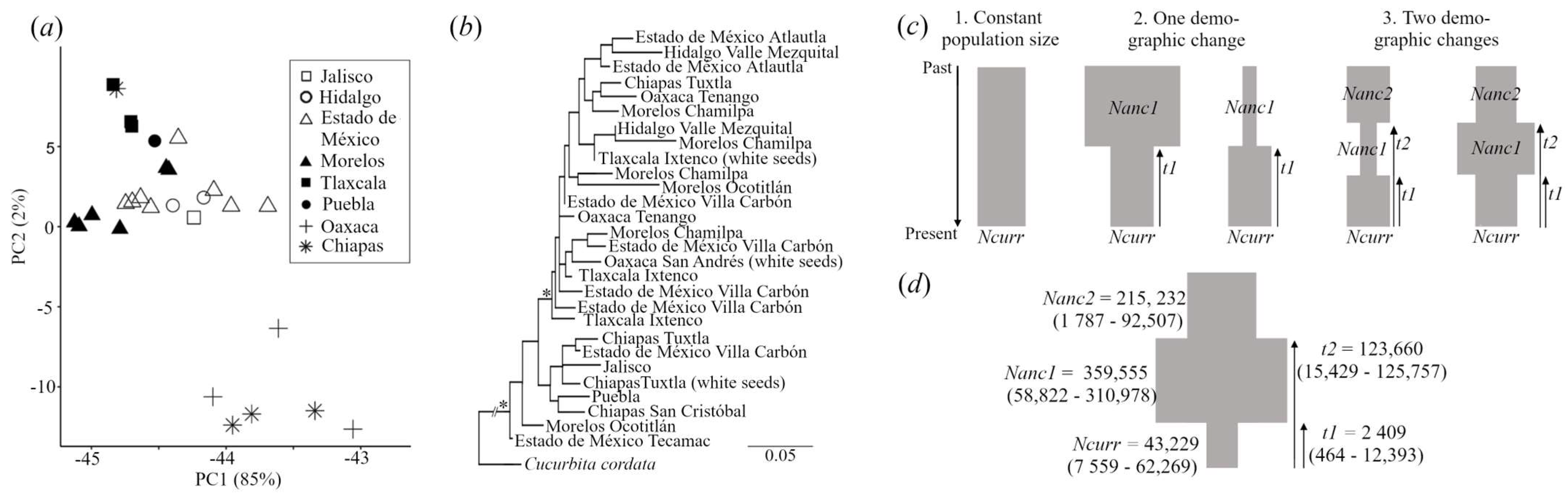
3.2. Cucurbita ficifolia Diversity and Demographics
3.3. Among-Taxa Differentiation and Gene Flow
4. Discussion
4.1. Diversity and History of C. ficifolia in Mexico
4.2. Gene Flow between C. ficifolia and Its Wild Relatives
4.3. Conservation of the Foetidissima Group
4.4. On the Nature of C. x scabridifolia
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Brown, A.H.D. Variation under Domestication in Plants: 1859 and Today. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Ross-Ibarra, J.; Morrell, P.L.; Gaut, B.S. Plant Domestication, a Unique Opportunity to Identify the Genetic Basis of Adaptation. Proc. Natl. Acad. Sci. USA 2007, 104 (Suppl. S1), 8641–8648. [Google Scholar] [CrossRef]
- Gregory, T.R. Artificial Selection and Domestication: Modern Lessons from Darwin’s Enduring Analogy. Evol. Educ. Outreach 2009, 2, 5–27. [Google Scholar] [CrossRef]
- Woodhouse, M.R.; Hufford, M.B. Parallelism and Convergence in Post-Domestication Adaptation in Cereal Grasses. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180245. [Google Scholar] [CrossRef]
- Renny-Byfield, S.; Wendel, J.F. Doubling down on Genomes: Polyploidy and Crop Plants. Am. J. Bot. 2014, 101, 1711–1725. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.S.; Purugganan, M.D. Evolution of Crop Species: Genetics of Domestication and Diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef] [PubMed]
- Gaut, B.S.; Seymour, D.K.; Liu, Q.; Zhou, Y. Demography and Its Effects on Genomic Variation in Crop Domestication. Nat. Plants 2018, 4, 512–520. [Google Scholar] [CrossRef]
- Gaut, B.S.; Díez, C.M.; Morrell, P.L. Genomics and the Contrasting Dynamics of Annual and Perennial Domestication. Trends Genet. 2015, 31, 709–719. [Google Scholar] [CrossRef]
- Ellstrand, N.C. Current Knowledge of Gene Flow in Plants: Implications for Transgene Flow. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 1163–1170. [Google Scholar] [CrossRef]
- Saban, J.M.; Romero, A.J.; Ezard, T.H.G.; Chapman, M.A. Extensive Crop-Wild Hybridization during Brassica Evolution and Selection during the Domestication and Diversification of Brassica Crops. Genetics 2023, 223, iyad027. [Google Scholar] [CrossRef]
- Ellstrand, N.C. Is Gene Flow the Most Important Evolutionary Force in Plants? Am. J. Bot. 2014, 101, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Hufford, M.B.; Berny Mier Y Teran, J.C.; Gepts, P. Crop Biodiversity: An Unfinished Magnum Opus of Nature. Annu. Rev. Plant Biol. 2019, 70, 727–751. [Google Scholar] [CrossRef] [PubMed]
- Dempewolf, H.; Hodgins, K.A.; Rummell, S.E.; Ellstrand, N.C.; Rieseberg, L.H. Reproductive Isolation during Domestication. Plant Cell 2012, 24, 2710–2717. [Google Scholar] [CrossRef] [PubMed]
- Allaby, R.G.; Stevens, C.J.; Kistler, L.; Fuller, D.Q. Emerging Evidence of Plant Domestication as a Landscape-Level Process. Trends Ecol. Evol. 2022, 37, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.J.; Gross, B.L. From Forest to Field: Perennial Fruit Crop Domestication. Am. J. Bot. 2011, 98, 1389–1414. [Google Scholar] [CrossRef] [PubMed]
- Zohary, D.; Hopf, M. Domestication of Plants in the Old World, 4th ed.; Oxford University Press: Oxford, UK, 2012; ISBN 978-0-19-968817-3. [Google Scholar]
- Allaby, R.G.; Fuller, D.Q.; Brown, T.A. The Genetic Expectations of a Protracted Model for the Origins of Domesticated Crops. Proc. Natl. Acad. Sci. USA 2008, 105, 13982–13986. [Google Scholar] [CrossRef]
- Harlan, J.R.; de Wet, J.M.J. Toward a Rational Classification of Cultivated Plants. Taxon 1971, 20, 509–517. [Google Scholar] [CrossRef]
- Cornille, A.; Gladieux, P.; Smulders, M.J.M.; Roldán-Ruiz, I.; Laurens, F.; Le Cam, B.; Nersesyan, A.; Clavel, J.; Olonova, M.; Feugey, L.; et al. New Insight into the History of Domesticated Apple: Secondary Contribution of the European Wild Apple to the Genome of Cultivated Varieties. PLoS Genet. 2012, 8, e1002703. [Google Scholar] [CrossRef]
- Choi, J.Y.; Platts, A.E.; Fuller, D.Q.; Hsing, Y.I.; Wing, R.A.; Purugganan, M.D.; Kim, Y. The Rice Paradox: Multiple Origins but Single Domestication in Asian Rice. Mol. Biol. Evol. 2017, 34, 969–979. [Google Scholar] [CrossRef]
- Wu, G.A.; Terol, J.; Ibanez, V.; López-García, A.; Pérez-Román, E.; Borredá, C.; Domingo, C.; Tadeo, F.R.; Carbonell-Caballero, J.; Alonso, R.; et al. Genomics of the Origin and Evolution of Citrus. Nature 2018, 554, 311–316. [Google Scholar] [CrossRef]
- Guerra-García, A.; Suárez-Atilano, M.; Mastretta-Yanes, A.; Delgado-Salinas, A.; Piñero, D. Domestication Genomics of the Open-Pollinated Scarlet Runner Bean (Phaseolus coccineus L.). Front. Plant Sci. 2017, 8, 1891. [Google Scholar] [CrossRef] [PubMed]
- Barrera-Redondo, J.; Sánchez-de la Vega, G.; Aguirre-Liguori, J.A.; Castellanos-Morales, G.; Gutiérrez-Guerrero, Y.T.; Aguirre-Dugua, X.; Aguirre-Planter, E.; Tenaillon, M.I.; Lira-Saade, R.; Eguiarte, L.E. The Domestication of Cucurbita argyrosperma as Revealed by the Genome of Its Wild Relative. Hortic. Res. 2021, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Letelier, A.; Aguirre-Liguori, J.A.; Piñero, D.; Vázquez-Lobo, A.; Eguiarte, L.E. The Relevance of Gene Flow with Wild Relatives in Understanding the Domestication Process. R. Soc. Open Sci. 2020, 7, 191545. [Google Scholar] [CrossRef] [PubMed]
- Razifard, H.; Ramos, A.; Della Valle, A.L.; Bodary, C.; Goetz, E.; Manser, E.J.; Li, X.; Zhang, L.; Visa, S.; Tieman, D.; et al. Genomic Evidence for Complex Domestication History of the Cultivated Tomato in Latin America. Mol. Biol. Evol. 2020, 37, 1118–1132. [Google Scholar] [CrossRef]
- Vavilov, N.I. Mexico and Central America as the Principal Centre of Origin of Cultivated Plants of the New World. Bull. Appl. Bot. Plant Breed. 1931, 26, 179–199. [Google Scholar]
- OECD Consensus Document on the Biology of Cucurbita L. (Squashes, Pumpkins, Zucchinis and Gourds); OECD: Paris, France, 2012.
- Castellanos-Morales, G.; Paredes-Torres, L.M.; Gámez, N.; Hernández-Rosales, H.S.; Sánchez-de la Vega, G.; Barrera-Redondo, J.; Aguirre-Planter, E.; Vázquez-Lobo, A.; Montes-Hernández, S.; Lira-Saade, R.; et al. Historical Biogeography and Phylogeny of Cucurbita: Insights from Ancestral Area Reconstruction and Niche Evolution. Mol. Phylogenet. Evol. 2018, 128, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Kates, H.R.; Soltis, P.S.; Soltis, D.E. Evolutionary and Domestication History of Cucurbita (Pumpkin and Squash) Species Inferred from 44 Nuclear Loci. Mol. Phylogenet. Evol. 2017, 111, 98–109. [Google Scholar] [CrossRef]
- Nee, M.H. The Domestication of Cucurbita (Cucurbitaceae). Econ. Bot. 1990, 44, 56–68. [Google Scholar] [CrossRef]
- Barrera-Redondo, J.; Hernández-Rosales, H.S.; Cañedo-Torres, V.; Aréstegui-Alegría, K.; Torres-Guevara, J.; Parra, F.; Torres-García, I.; Casas, A. Landrace Diversity and Local Selection Criteria of Domesticated Squashes and Gourds (Cucurbita) in the Central Andean Mountain Range of Peru: Tomayquichua, Huánuco. Bot. Sci. 2020, 98, 101–116. [Google Scholar] [CrossRef]
- Andres, T.C. Biosystematics, Theories on the Origin, and Breeding Potential of Cucurbita ficifolia. In Biology and Utilization of the Cucurbitaceae; Bates, D.M., Robinson, R.W., Jeffreys, C., Eds.; Cornell University Library: New York, NY, USA, 1990; pp. 102–119. [Google Scholar]
- Cerón González, L.; Legaria Solano, J.P.; Villanueva Verduzco, C.; Sahagún Castellanos, J. Diversidad Genética En Cuatro Especies Mexicanas de Calabaza (Cucurbita spp.). Rev. Fitotec. Mex. 2010, 33, 189–196. [Google Scholar] [CrossRef]
- Rice, A.; Glick, L.; Abadi, S.; Einhorn, M.; Kopelman, N.M.; Salman-Minkov, A.; Mayzel, J.; Chay, O.; Mayrose, I. The Chromosome Counts Database (CCDB)—A Community Resource of Plant Chromosome Numbers. New Phytol. 2015, 206, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Lira-Saade, R. Estudios Taxonómicos y Ecogeográficos de Las Cucurbitaceae Latinoamericanas de Importancia Económica; International Plant Genetic Resources Institute: Rome, Italy, 1995; ISBN 92-9043-263-2. [Google Scholar]
- Piperno, D.R. The Origins of Plant Cultivation and Domestication in the New World Tropics. Curr. Anthropol. 2011, 52, S453–S470. [Google Scholar] [CrossRef]
- Whitaker, T.W. Archeological Cucurbits. Econ. Bot. 1981, 35, 460–466. [Google Scholar] [CrossRef]
- Whitaker, T.W.; Cutler, H.C. Pre-Historic Cucurbits from the Valley of Oaxaca. Econ. Bot. 1971, 25, 123–127. [Google Scholar] [CrossRef]
- Andres, T.C. Relationship of Cucurbita scabridifolia to C. foetidissima and C. pedatifolia: A Case of Natural Interspecific Hybridization. Cucurbit Genet. Coop. Rep. 1987, 10, 74–75. [Google Scholar]
- Montero-Pau, J.; Blanca, J.; Bombarely, A.; Ziarsolo, P.; Esteras, C.; Martí-Gómez, C.; Ferriol, M.; Gómez, P.; Jamilena, M.; Mueller, L.; et al. De Novo Assembly of the Zucchini Genome Reveals a Whole-Genome Duplication Associated with the Origin of the Cucurbita Genus. Plant Biotechnol. J. 2018, 16, 1161–1171. [Google Scholar] [CrossRef]
- Khoury, C.K.; Carver, D.; Kates, H.R.; Achicanoy, H.A.; van Zonneveld, M.; Thomas, E.; Heinitz, C.; Jarret, R.; Labate, J.A.; Reitsma, K.; et al. Distributions, Conservation Status, and Abiotic Stress Tolerance Potential of Wild Cucurbits (Cucurbita L.). Plants People Planet 2019, 2, 269–283. [Google Scholar] [CrossRef]
- Andres, T.C. Hybridization of Cucurbita foetidissima with C. pedatifolia, C. radicans, and C. ficifolia. Cucurbit Genet. Coop. Rep. 1987, 10, 72–73. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De Novo Assembly of Organelle Genomes from Whole Genome Data. Nucleic Acids Res. 2016, 45, gkw955. [Google Scholar] [CrossRef]
- Barrera-Redondo, J.; Ibarra-Laclette, E.; Vázquez-Lobo, A.; Gutiérrez-Guerrero, Y.T.; Sánchez de la Vega, G.; Piñero, D.; Montes-Hernández, S.; Lira-Saade, R.; Eguiarte, L.E. The Genome of Cucurbita argyrosperma (Silver-Seed Gourd) Reveals Faster Rates of Protein-Coding Gene and Long Noncoding RNA Turnover and Neofunctionalization within Cucurbita. Mol. Plant 2019, 12, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the Evolution of Mitochondrial Genome Size from Complete Sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H.; et al. Efficient de Novo Assembly of Highly Heterozygous Genomes from Whole-Genome Shotgun Short Reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Sam Ma, Z. DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef] [PubMed]
- Alonge, M.; Soyk, S.; Ramakrishnan, S.; Wang, X.; Goodwin, S.; Sedlazeck, F.J.; Lippman, Z.B.; Schatz, M.C. RaGOO: Fast and Accurate Reference-Guided Scaffolding of Draft Genomes. Genome Biol. 2019, 20, 224. [Google Scholar] [CrossRef]
- Sun, H.; Wu, S.; Zhang, G.; Jiao, C.; Guo, S.; Ren, Y.; Zhang, J.; Zhang, H.; Gong, G.; Jia, Z.; et al. Karyotype Stability and Unbiased Fractionation in the Paleo-Allotetraploid Cucurbita Genomes. Mol. Plant 2017, 10, 1293–1306. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Eaton, D.A.R.; Overcast, I. Ipyrad: Interactive Assembly and Analysis of RADseq Datasets. Bioinformatics 2020, 36, 2592–2594. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.; Lunter, G.; Marth, G.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559. [Google Scholar] [CrossRef] [PubMed]
- Kistler, L.; Newsom, L.A.; Ryan, T.M.; Clarke, A.C.; Smith, B.D.; Perry, G.H. Gourds and Squashes (Cucurbita spp.) Adapted to Megafaunal Extinction and Ecological Anachronism through Domestication. Proc. Natl. Acad. Sci. USA 2015, 112, 15107–15112. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Dugua, X.; Castellanos-Morales, G.; Paredes-Torres, L.M.; Hernández-Rosales, H.S.; Barrera-Redondo, J.; Sánchez-de la Vega, G.; Tapia-Aguirre, F.; Ruiz-Mondragón, K.Y.; Scheinvar, E.; Hernández, P.; et al. Evolutionary Dynamics of Transferred Sequences between Organellar Genomes in Cucurbita. J. Mol. Evol. 2019, 87, 327–342. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The Interpretation of Population Structure by F Statistics with Special Regard to Systems of Mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An Analysis Tool Set for Population Genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Gutenkunst, R.N.; Hernandez, R.D.; Williamson, S.H.; Bustamante, C.D. Inferring the Joint Demographic History of Multiple Populations from Multidimensional SNP Frequency Data. PLoS Genet. 2009, 5, e1000695. [Google Scholar] [CrossRef]
- Excoffier, L.; Dupanloup, I.; Huerta-Sanchez, E.; Sousa, V.C.; Foll, M. Robust Demographic Inference from Genomic and SNP Data. PLoS Genet. 2013, 9, e1003905. [Google Scholar] [CrossRef]
- Burnham, K.P.; Anderson, R.P. Multimodel Inference: Understanding AIC and BIC in Model Selection. Sociol. Methods Res. 2004, 33, 261–304. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population Growth Makes Waves in the Distribution of Pairwise Genetic Differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Excoffier, L. Estimation of Past Demographic Parameters from the Distribution of Pairwise Differences When the Mutation Rates Vary among Sites: Application to Human Mitochondrial DNA. Genetics 1999, 152, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Durand, E.; Patterson, N.; Reich, D.; Slatkin, M. Testing for Ancient Admixture between Closely Related Populations. Mol. Biol. Evol. 2011, 28, 2239–2252. [Google Scholar] [CrossRef]
- Malinsky, M.; Matschiner, M.; Svardal, H. Dsuite—Fast D-Statistics and Related Admixture Evidence from VCF Files. Mol. Ecol. Resour. 2021, 21, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Martínez-González, C.; Castellanos-Morales, G.; Barrera-Redondo, J.; Sánchez-de la Vega, G.; Hernández-Rosales, H.S.; Gasca-Pineda, J.; Aguirre-Planter, E.; Moreno-Letelier, A.; Escalante, A.E.; Montes-Hernández, S.; et al. Recent and Historical Gene Flow in Cultivars, Landraces, and a Wild Taxon of Cucurbita pepo in Mexico. Front. Ecol. Evol. 2021, 9, 656051. [Google Scholar] [CrossRef]
- Hernández-Rosales, H.S.; Castellanos-Morales, G.; Sánchez-de la Vega, G.; Aguirre-Planter, E.; Montes-Hernández, S.; Lira-Saade, R.; Eguiarte, L.E. Phylogeographic and Population Genetic Analyses of Cucurbita moschata Reveal Divergence of Two Mitochondrial Lineages Linked to an Elevational Gradient. Am. J. Bot. 2020, 107, 510–525. [Google Scholar] [CrossRef]
- Hurd, P.D.J.; Linsley, E.G.; Whitaker, T.W. Squash and Gourd Bees (Peponapis, Xenoglossa) and the Origin of the Cultivated Cucurbita. Evolution 1971, 25, 218–234. [Google Scholar]
- Smith, B.D. The Initial Domestication of Cucurbita pepo in the Americas 10,000 Years Ago. Science 1997, 276, 932–934. [Google Scholar] [CrossRef]
- Ranere, A.J.; Piperno, D.R.; Holst, I.; Dickau, R.; Iriarte, J. The Cultural and Chronological Context of Early Holocene Maize and Squash Domestication in the Central Balsas River Valley, Mexico. Proc. Natl. Acad. Sci. USA 2009, 106, 5014–5018. [Google Scholar] [CrossRef]
- Larson, G. Genetics and Domestication. Curr. Anthropol. 2011, 52, S485–S495. [Google Scholar] [CrossRef]
- Giannini, T.C.; Lira-Saade, R.; Saraiva, A.M.; Alves-dos-Santos, I. Ecological Niche Similarities of Peponapis Bees and Non-Domesticated Cucurbita Species. Ecol. Model. 2011, 222, 2011–2018. [Google Scholar] [CrossRef]
- OECD Safety Assessment of Transgenic Organisms in the Environment; OECD: Paris, France, 2016; Volume 5.
- Sánchez-de la Vega, G.; Castellanos-Morales, G.; Gámez, N.; Hernández-Rosales, H.S.; Vázquez-Lobo, A.; Aguirre-Planter, E.; Jaramillo-Correa, J.P.; Montes-Hernández, S.; Lira-Saade, R.; Eguiarte, L.E. Genetic Resources in the “Calabaza Pipiana” Squash (Cucurbita argyrosperma) in Mexico: Genetic Diversity, Genetic Differentiation and Distribution Models. Front. Plant Sci. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Purugganan, M.D.; Fuller, D.Q. The Nature of Selection during Plant Domestication. Nature 2009, 457, 843–848. [Google Scholar] [CrossRef]
- Šiško, M.; Ivančič, A.; Bohanec, B. Genome Size Analysis in the Genus Cucurbita and Its Use for Determination of Interspecific Hybrids Obtained Using the Embryo-Rescue Technique. Plant Sci. 2003, 165, 663–669. [Google Scholar] [CrossRef]
- Guo, J.; Xu, W.; Hu, Y.; Huang, J.; Zhao, Y.; Zhang, L.; Huang, C.H.; Ma, H. Phylotranscriptomics in Cucurbitaceae Reveal Multiple Whole-Genome Duplications and Key Morphological and Molecular Innovations. Mol. Plant 2020, 13, 1117–1133. [Google Scholar] [CrossRef]
- Weiling, F. Genomanalytische Untersuchungen Bei Kürbis (Cucurbita L.). Zuchter 1959, 29, 161–179. [Google Scholar] [CrossRef]
- Rieseberg, L.H.; Blackman, B.K. Speciation Genes in Plants. Ann. Bot. 2010, 106, 439–455. [Google Scholar] [CrossRef]
- Papa, R. Gene Flow between Crops and Their Wild Relatives. In Proceedings of the International Workshop on the Role of Biotechnology for the Characterization and Conservation of Crop Forestry, Animal and Fishery Genetic Resources, Turin, Italy, 5–7 March 2005; pp. 71–76. [Google Scholar]
- Ellstrand, N.C.; Meirmans, P.; Rong, J.; Bartsch, D.; Ghosh, A.; de Jong, T.J.; Haccou, P.; Lu, B.-R.; Snow, A.A.; Neal Stewart, C.; et al. Introgression of Crop Alleles into Wild or Weedy Populations. Annu. Rev. Ecol. Evol. Syst. 2013, 44, 325–345. [Google Scholar] [CrossRef]
- Todesco, M.; Pascual, M.A.; Owens, G.L.; Ostevik, K.L.; Moyers, B.T.; Hübner, S.; Heredia, S.M.; Hahn, M.A.; Caseys, C.; Bock, D.G.; et al. Hybridization and Extinction. Evol. Appl. 2016, 9, 892–908. [Google Scholar] [CrossRef]
- Lira, R.; Téllez, O.; Dávila, P. The Effects of Climate Change on the Geographic Distribution of Mexican Wild Relatives of Domesticated Cucurbitaceae. Genet. Resour. Crop Evol. 2009, 56, 691–703. [Google Scholar] [CrossRef]
- IUCN. The IUCN Red List of Threatened Species. Version 2022-2. Available online: https://www.iucnredlist.org (accessed on 15 November 2022).
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef]
- Magallón, S.; Gómez-Acevedo, S.; Sánchez-Reyes, L.L.; Hernández-Hernández, T. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 2015, 207, 437–453. [Google Scholar] [CrossRef]
- Overcast, I. 2020 easySFS.py. Available online: https://github.com/isaacovercast/easySFS (accessed on 17 September 2023).
- Hamilton, M.B. Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Mol. Ecol. 1999, 8, 521–523. [Google Scholar]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Status | Habitat | Number of Individuals | Number of Collection Sites |
|---|---|---|---|---|
| C. foetidissima | Wild | Xerophytic | 17 | 12 |
| C. x scabridifolia | Wild | Xerophytic | 19 | 7 |
| C. pedatifolia | Wild | Xerophytic | 34 | 12 |
| C. radicans | Wild | Xerophytic | 44 | 14 |
| C. ficifolia | Domesticated | Mesophytic | 36 | 15 |
| C. cordata (outgroup) | Wild | Xerophytic | 11 | 3 |
| Taxon | π (var) | HE (var) | HO (var) | FIS (var) | Reference |
|---|---|---|---|---|---|
| Cucurbita ficifolia | 0.225 (0.025) | 0.226 (0.024) | 0.148 (0.016) | 0.233 (0.054) | This study |
| C. pepo subsp. pepo | 0.197 (0.026) | 0.196 (0.025) | 0.169 (0.023) | 0.116 (0.097) | [70] |
| C. argyrosperma subsp. argyrosperma | 0.095 (0.010) | 0.094 (0.012) | 0.094 (0.012) | 0.034 (0.030) | [23] |
| Model | Historical Demography | Ln MaxEstLhood | Nparams | AIC | ΔAIC | Rank |
|---|---|---|---|---|---|---|
| 1 | Constant population size | −5144.763 | 1 | 10,291.53 | −3.48 | 3 |
| 2 | One demographic change | −5142.073 | 3 | 10,290.15 | −2.1 | 2 |
| 3 | Two demographic changes | −5139.025 | 5 | 10,288.05 | 0 | 1 |
| Model | Gene Flow between C. ficifolia and Wild Relatives | Ln MaxEstLhood | Nparams | AIC | ΔAIC | Rank |
|---|---|---|---|---|---|---|
| I.1 | Absent | −53,713.31 | 13 | 107,452.6 | 4883.5 | 5 |
| I.2 | C. ficifolia- C. foetidissima | −52,057.21 | 15 | 104,144.4 | 1575.3 | 3 |
| I.3 | C. ficifolia- C. x scabridifolia | −52,272.53 | 15 | 104,575.1 | 2006.0 | 4 |
| I.4 | C. ficifolia- C. pedatifolia | −51,958.6 | 15 | 103,947.2 | 1378.1 | 2 |
| I.5 | C. ficifolia- C. radicans | −51,269.55 | 15 | 102,569.1 | 0 | 1 |
| Trio | BBAA Frequency | ABBA Frequency | BABA Frequency | D-Statistic | Z-Score | p-Value | ||
|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | ||||||
| pedatifolia | foetscabri | ficifolia | 4.43434 | 3.64444 | 3.55652 | 0.0122084 | 0.804987 | 0.2104 ns |
| radicans | foetscabri | ficifolia | 3.92942 | 3.61323 | 3.51278 | 0.0140957 | 0.845496 | 0.1989 ns |
| radicans | pedatifolia | ficifolia | 4.35583 | 3.87077 | 3.85665 | 0.00182715 | 0.109742 | 0.4563 ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguirre-Dugua, X.; Barrera-Redondo, J.; Gasca-Pineda, J.; Vázquez-Lobo, A.; López-Camacho, A.; Sánchez-de la Vega, G.; Castellanos-Morales, G.; Scheinvar, E.; Aguirre-Planter, E.; Lira-Saade, R.; et al. Population Genomics of Domesticated Cucurbita ficifolia Reveals a Recent Bottleneck and Low Gene Flow with Wild Relatives. Plants 2023, 12, 3989. https://doi.org/10.3390/plants12233989
Aguirre-Dugua X, Barrera-Redondo J, Gasca-Pineda J, Vázquez-Lobo A, López-Camacho A, Sánchez-de la Vega G, Castellanos-Morales G, Scheinvar E, Aguirre-Planter E, Lira-Saade R, et al. Population Genomics of Domesticated Cucurbita ficifolia Reveals a Recent Bottleneck and Low Gene Flow with Wild Relatives. Plants. 2023; 12(23):3989. https://doi.org/10.3390/plants12233989
Chicago/Turabian StyleAguirre-Dugua, Xitlali, Josué Barrera-Redondo, Jaime Gasca-Pineda, Alejandra Vázquez-Lobo, Andrea López-Camacho, Guillermo Sánchez-de la Vega, Gabriela Castellanos-Morales, Enrique Scheinvar, Erika Aguirre-Planter, Rafael Lira-Saade, and et al. 2023. "Population Genomics of Domesticated Cucurbita ficifolia Reveals a Recent Bottleneck and Low Gene Flow with Wild Relatives" Plants 12, no. 23: 3989. https://doi.org/10.3390/plants12233989