Complete Chloroplast Genomes of Chlorophytum comosum and Chlorophytum gallabatense: Genome Structures, Comparative and Phylogenetic Analysis


 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
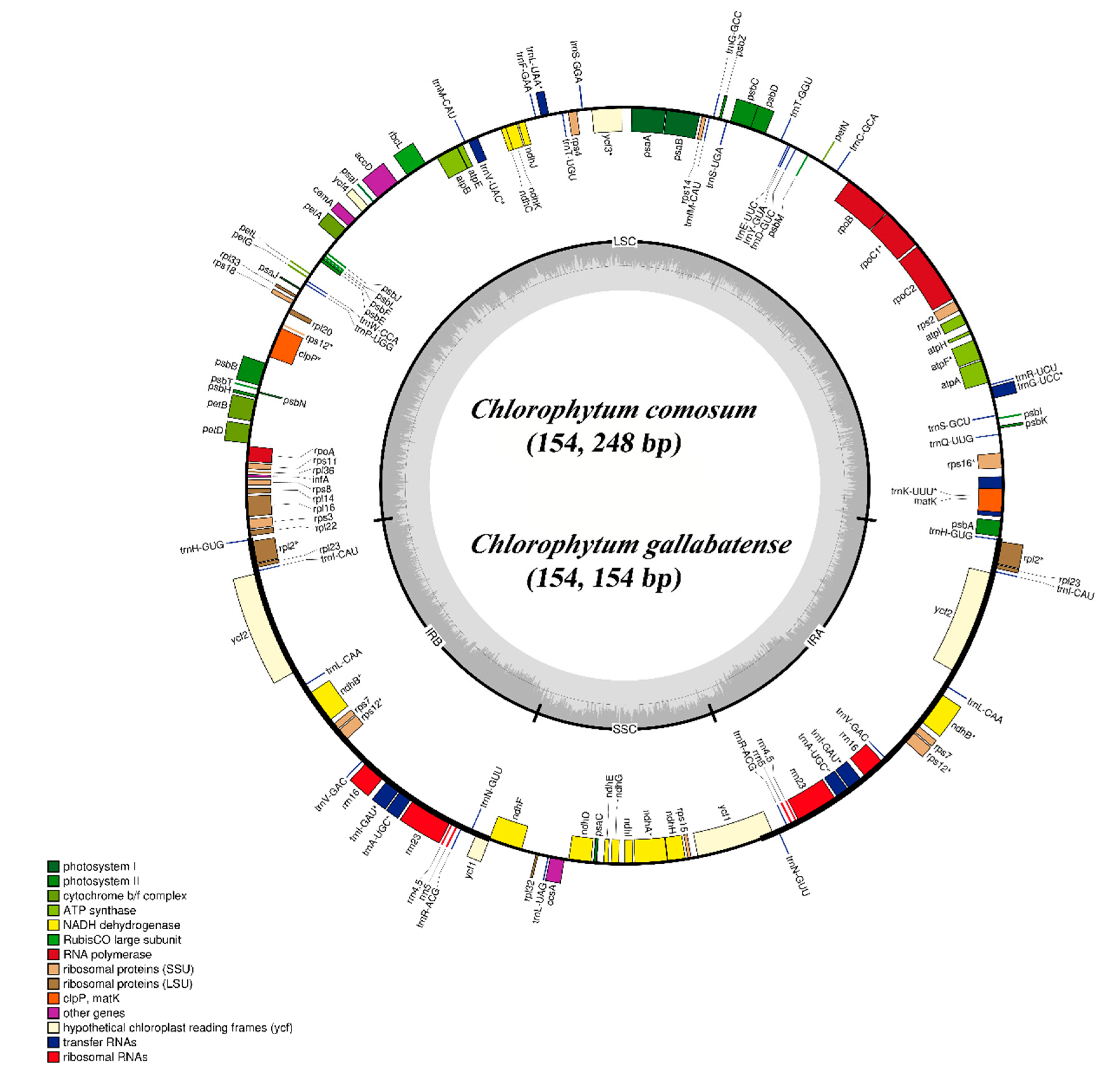
2.1. C. comosum and C. gallabatense Chloroplast Genome Features
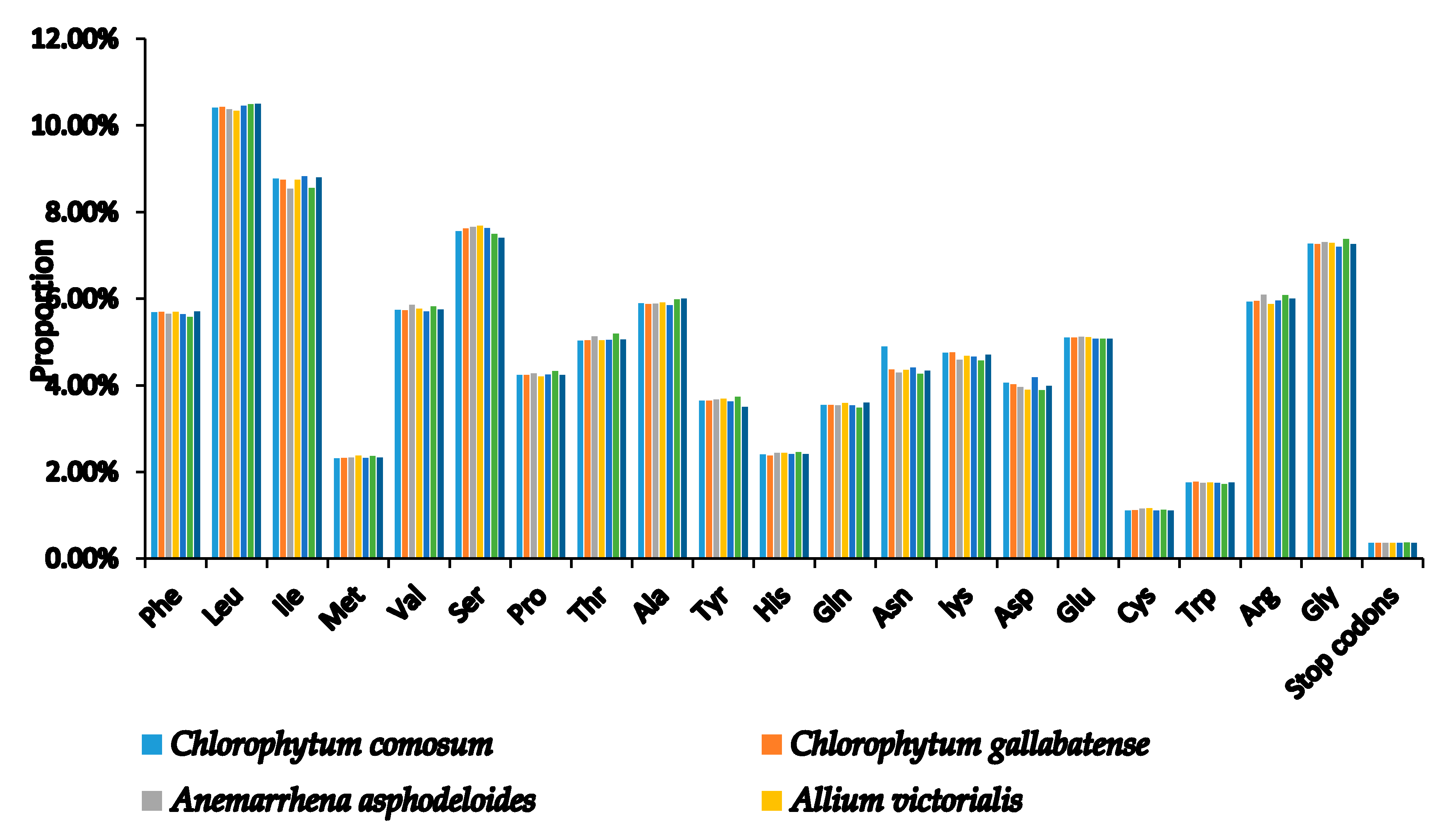
2.2. Codon Usage Analysis
2.3. RNA Editing Sites
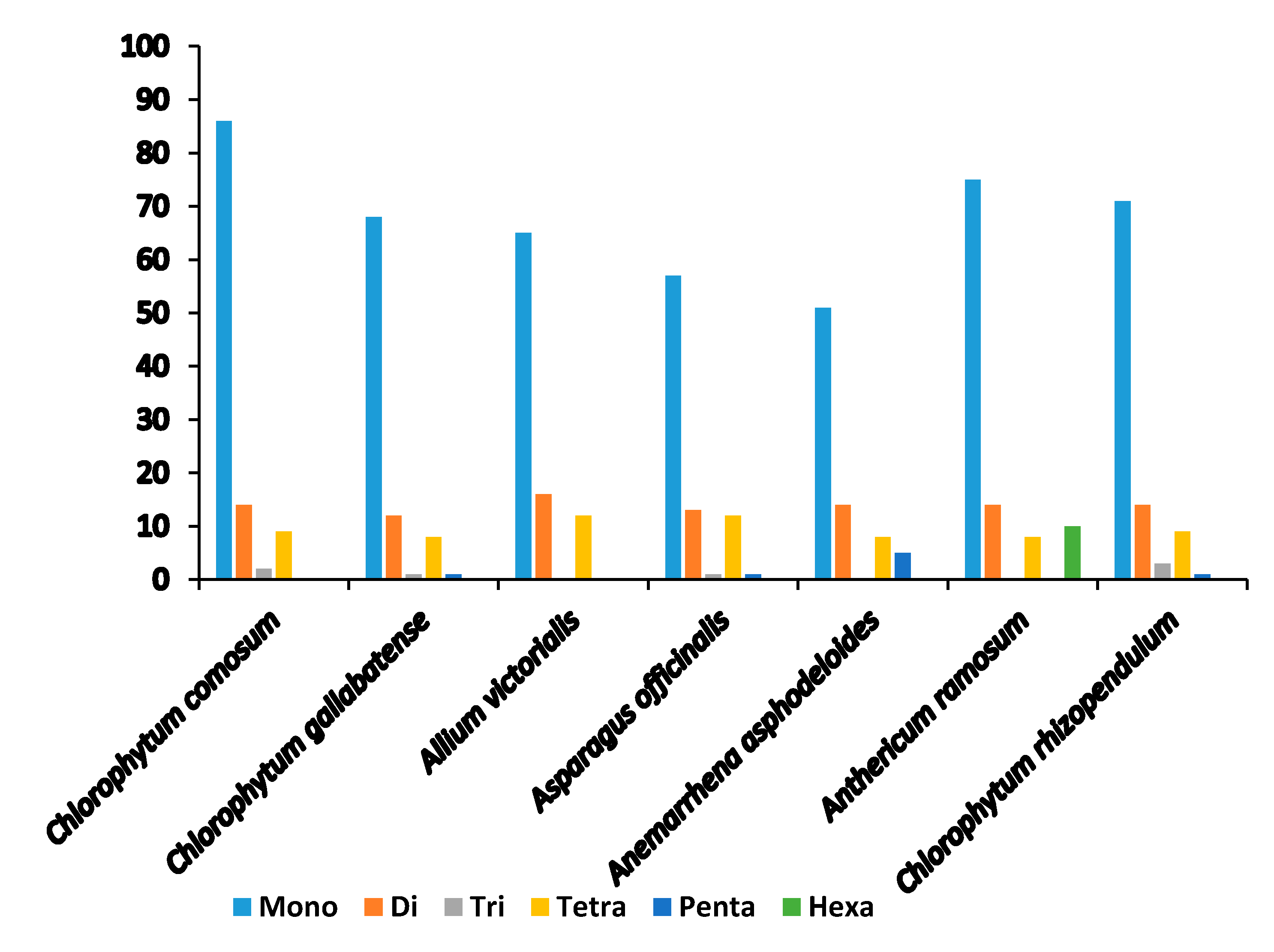
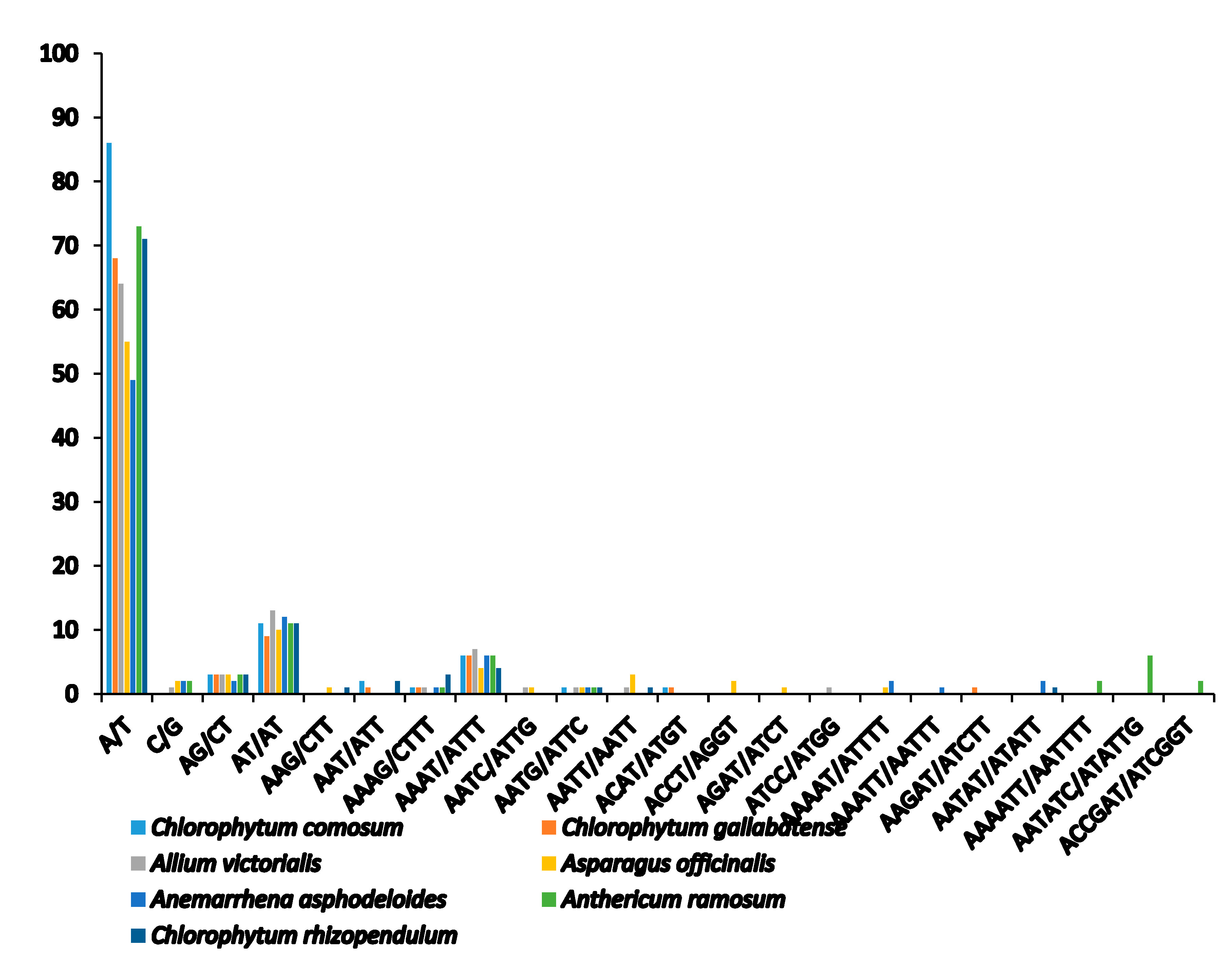
2.4. Analysis of SSRs
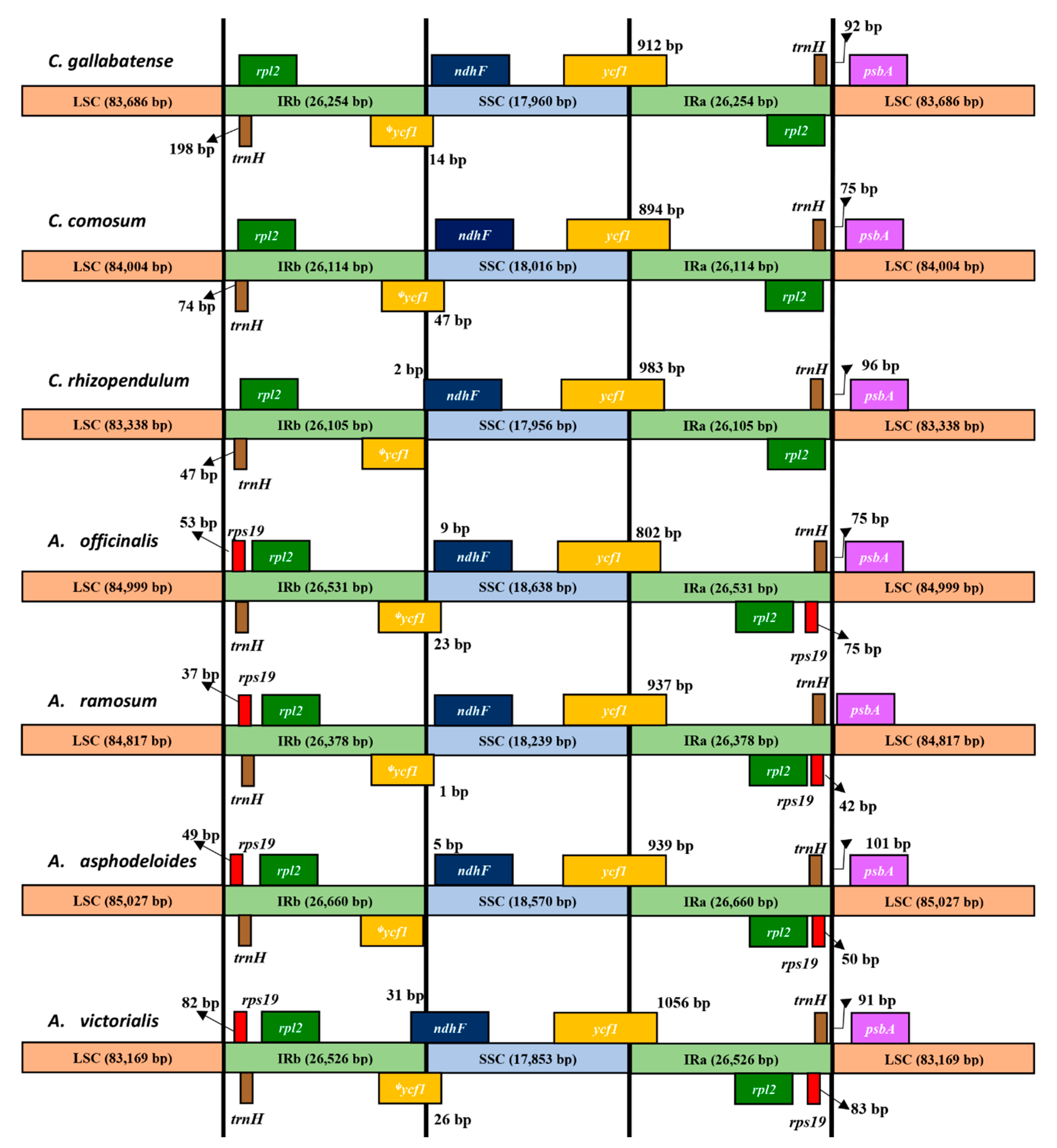
2.5. IR Expansion and Contraction
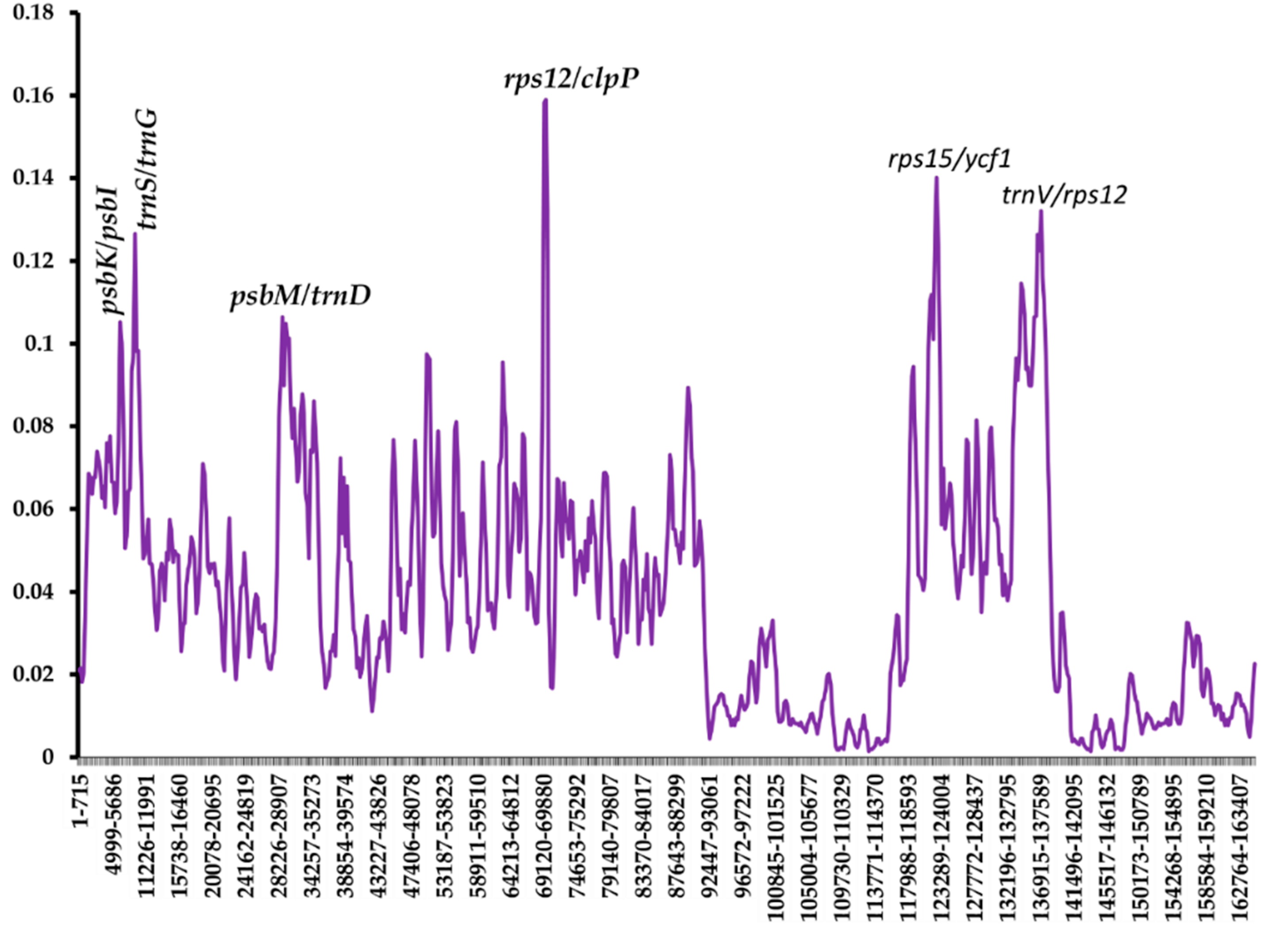
2.6. Analysis of Nucleotide Diversity
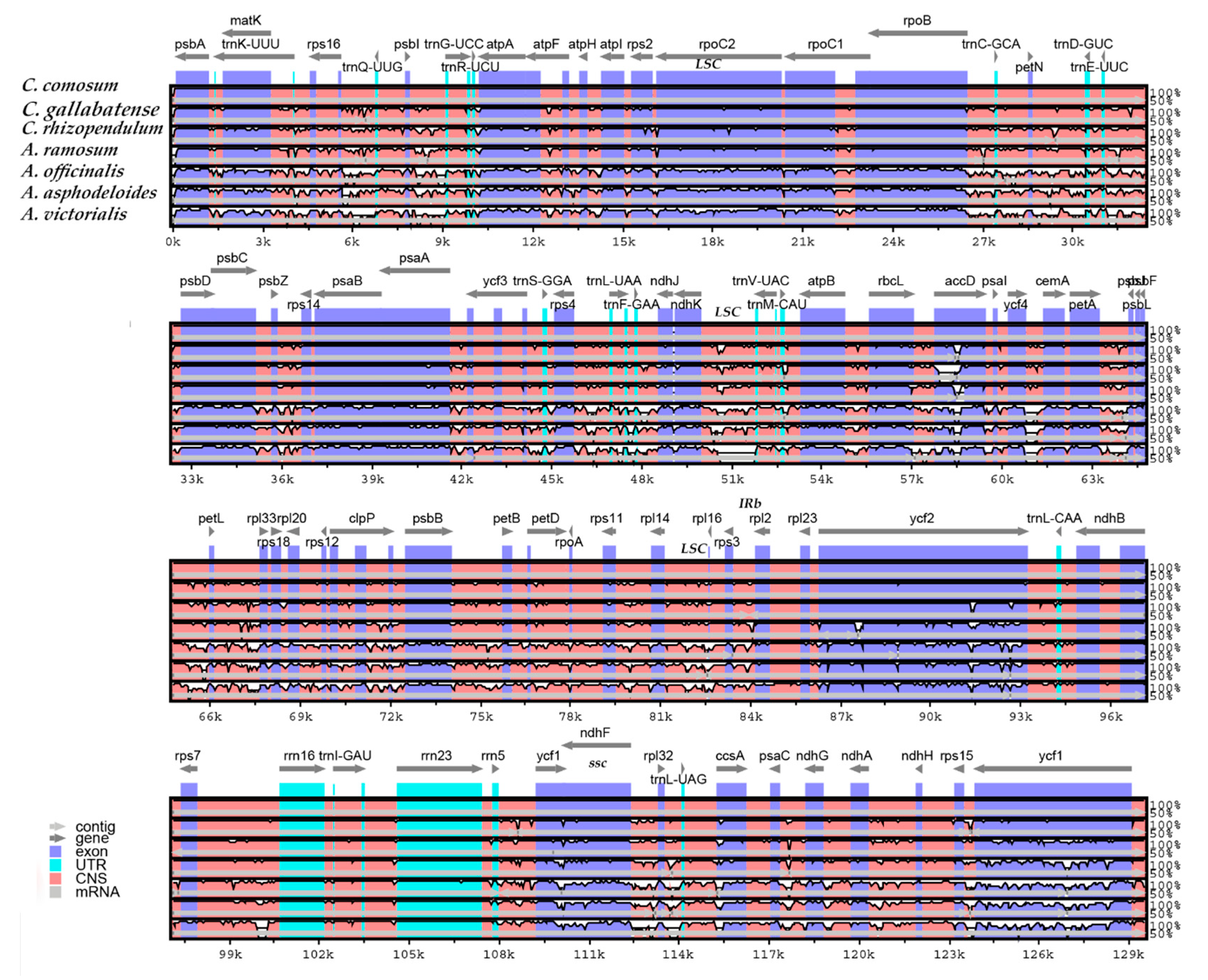
2.7. Comparative Genome Analysis
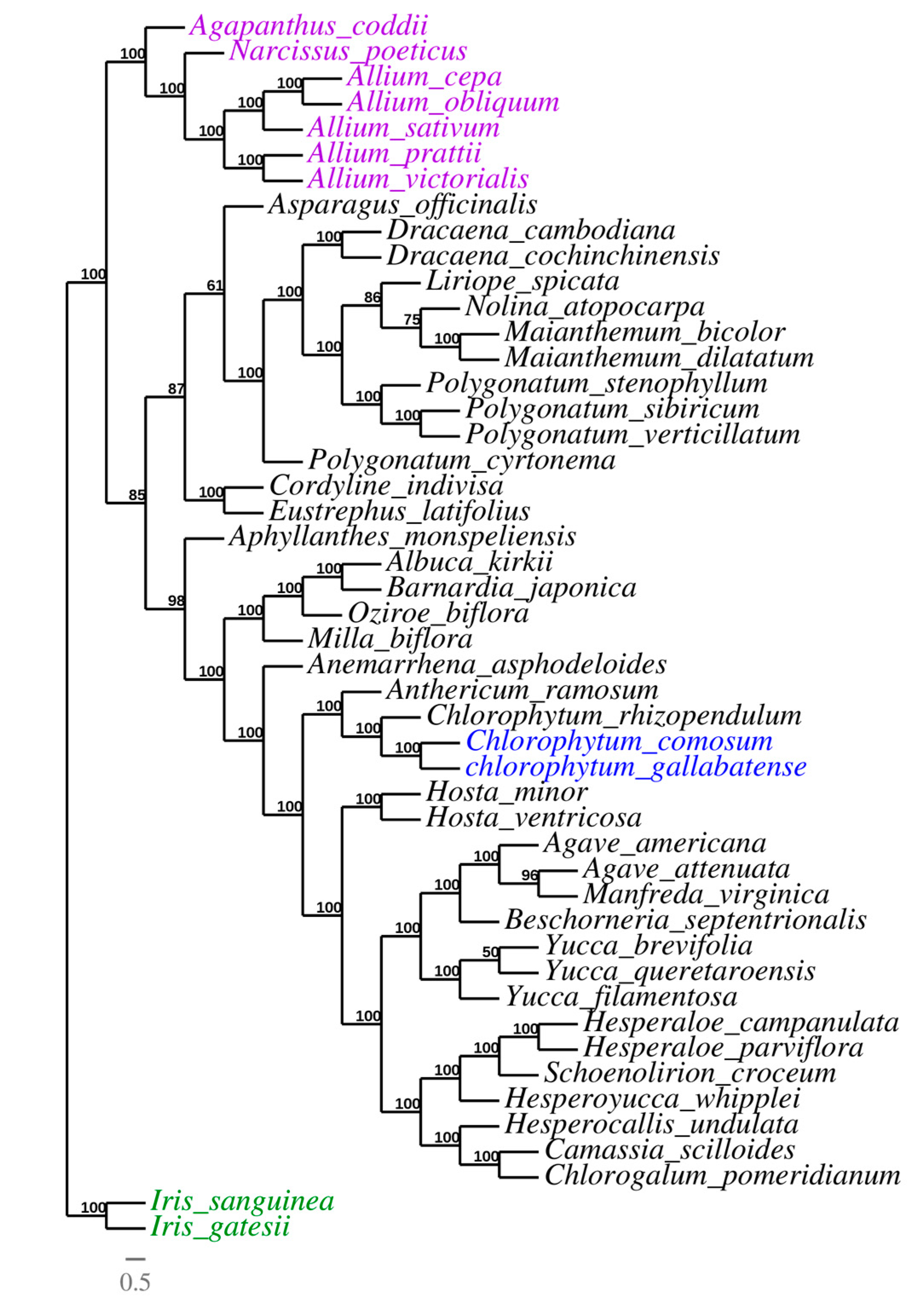
2.8. Phylogenetic Analysis
3. Materials and Methods
3.1. DNA Isolation and Chloroplast Genome Sequencing
3.2. Genome Assembly and Annotation
3.3. Genome Structure and Comparison
3.4. Codon Usage
3.5. Simple Sequence Repeats
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Basu, S.; Jha, T.B. Cytogenetic studies in four species of Chlorophytum Ker-Gawl. (Liliaceae). The Nucleus 2011, 54, 123–132. [Google Scholar] [CrossRef]
- Govaerts, R.; Zonneveld, B.; Zona, S. World Checklist of Asparagaceae; Facilitated by the Royal Botanic Gardens: Kew, UK, 2012; Available online: http://apps.kew.org/wcsp/ (accessed on 26 February 2020).
- Malpure, N.V.; Yadav, S. Chlorophytum gothanense, a new species of Anthericaceae from the Western Ghats of India. Kew Bull. 2009, 64, 739–741. [Google Scholar] [CrossRef]
- Govaerts, R. World Checklist of Selected Plant Families Asparagaceae; Royal Botanic Gardens: Kew, UK, 2015. [Google Scholar]
- Lekhak, M.; Adsul, A.; Yadav, S. Cytotaxonomical investigations into the genus Chlorophytum from India. Kew Bull. 2012, 67, 285–292. [Google Scholar] [CrossRef]
- Group, A.P. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar]
- Adsul, A.A. Taxonomic Revision of Genus Chlorophytum Ker Gawl for India. Ph.D. Thesis, Shivaji University, Kolhapur India, 2015. [Google Scholar]
- Kumar, M.; Meena, P.; Verma, S.; Kumar, M.; Kumar, A. Anti-tumour, anti-mutagenic and chemomodulatory potential of Chlorophytum borivilianum. Asian Pac. J. Cancer Prev. 2010, 11, 327–334. [Google Scholar]
- Kalra, S.; Kumar, S.; Lakhanpal, N.; Kaur, J.; Singh, K. Characterization of squalene synthase gene from Chlorophytum borivilianum (Sant. and Fernand.). Mol. Biotechnol. 2013, 54, 944–953. [Google Scholar] [CrossRef]
- Deore, S.; Khadabadi, S. Standardisation and pharmaceutical evaluation of Chlorophytum borivilianum mucilage. Rasayan J. Chem. 2008, 1, 887–892. [Google Scholar]
- Khanam, Z.; Singh, O.; Singh, R.; Bhat, I.U.H. Safed musli (Chlorophytum borivilianum): A review of its botany, ethnopharmacology and phytochemistry. J. Ethnopharmacol. 2013, 150, 421–441. [Google Scholar] [CrossRef]
- Kaushik, N. Saponins of Chlorophytum species. Phytochem. Rev. 2005, 4, 191–196. [Google Scholar] [CrossRef]
- Haque, R.; Saha, S.; Bera, T. A peer reviewed literature on medicinal activity of Chlorophytum borivilianum commercial medicinal plant. Int. J. Drug Dev. Res. 2011, 3, 1116, 1130. [Google Scholar]
- O’Donnell, G.; Bucar, F.; Gibbons, S. Phytochemistry and antimycobacterial activity of Chlorophytum inornatum. Phytochemistry 2006, 67, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Geetha, K. Characterization, genetic improvement and cultivation of Chlorophytum borivilianum—an important medicinal plant of India. Plant Genet. Resour. 2005, 3, 264–272. [Google Scholar] [CrossRef]
- Adsul, A.A.; Lekhak, M.M.; Yadav, S.R. Chlorophytum sharmae (Asparagaceae): A new species from Kerala, India. Kew Bull. 2014, 69. [Google Scholar] [CrossRef]
- Bjorå, C.S.; Hemp, A.; Hoell, G.; Nordal, I. A taxonomic and ecological analysis of two forest Chlorophytum taxa (Anthericaceae) on Mount Kilimanjaro, Tanzania. Plant Syst. Evol. 2008, 274, 243–253. [Google Scholar] [CrossRef]
- McKain, M.R.; McNeal, J.R.; Kellar, P.R.; Eguiarte, L.E.; Pires, J.C.; Leebens-Mack, J. Timing of rapid diversification and convergent origins of active pollination within Agavoideae (Asparagaceae). Am. J. Bot. 2016, 103, 1717–1729. [Google Scholar] [CrossRef]
- Deguilloux, M.-F.; Pemonge, M.-H.l.N.; Petit, R.M.J. Use of chloroplast microsatellites to differentiate oak populations. Ann. For. Sci. 2004, 61, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, H.; Emes, M. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, J.; Li, W.; Zhang, X.; Xu, Y.; Xu, F.; Zhu, H.; Wang, X. The complete chloroplast genome of Euphrasia regelii, pseudogenization of ndh genes and the phylogenetic relationships within Orobanchaceae. Front. Genet. 2019, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Bendich, A.J. Circular chloroplast chromosomes: The grand illusion. Plant Cell 2004, 16, 1661–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium× hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-P.; Huang, J.-P.; Wu, C.-S.; Hsu, C.-Y.; Chaw, S.-M. Comparative chloroplast genomics reveals the evolution of Pinaceae genera and subfamilies. Genome Biol. Evol. 2010, 2, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [Green Version]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Yi, X.; Yang, Y.-X.; Su, Y.-J.; Wang, T. Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: Insights into evolutionary changes in fern chloroplast genomes. BMC Evol. Biol. 2009, 9, 130. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Li, Y.; Cai, Q.; Lin, F.; Huang, P.; Zheng, Y. Development of chloroplast genomic resources for Akebia quinata (Lardizabalaceae). Conserv. Genet. Resour. 2016, 8, 447–449. [Google Scholar] [CrossRef]
- Sun, M.; Li, J.; Li, D.; Shi, L. Complete chloroplast genome sequence of the medical fern Drynaria roosii and its phylogenetic analysis. Mitochondrial Dna Part B 2017, 2, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Chai, X.; Rao, Y.; Tu, X.; Du, S. The Complete Chloroplast Genome Sequence of Asparagus (Asparagus officinalis L.) and its phylogenetic positon within Asparagales. Int. J. Plant Biol. Res. 2017, 5, 121–128. [Google Scholar]
- Cui, Y.; Nie, L.; Sun, W.; Xu, Z.; Wang, Y.; Yu, J.; Song, J.; Yao, H. Comparative and Phylogenetic Analyses of Ginger (Zingiber officinale) in the Family Zingiberaceae Based on the Complete Chloroplast Genome. Plants 2019, 8, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iram, S.; Hayat, M.Q.; Tahir, M.; Gul, A.; Ahmed, I. Chloroplast Genome Sequence of Artemisia scoparia: Comparative Analyses and Screening of Mutational Hotspots. Plants 2019, 8, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Feng, D.; Song, G.; Wei, X.; Chen, L.; Wu, X.; Li, X.; Zhu, Z. The first intron of rice EPSP synthase enhances expression of foreign gene. Sci. China Ser. C: Life Sci. 2003, 46, 561. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, Z.; Huang, S.; An, W.; Li, J.; Zheng, X. Comprehensive Analysis of Rhodomyrtus tomentosa Chloroplast Genome. Plants 2019, 8, 89. [Google Scholar] [CrossRef] [Green Version]
- Meerow, A.W. Convergence or reticulation? Mosaic evolution in the canalized American Amaryllidaceae. In Diversity, Phylogeny and Evolution in the Monocotyledons; Seberg, O., Petersen, G., Barfod, A.S., Davis, J.I., Eds.; Aarhus University Press: Aarhus, Denmark, 2010; pp. 145–168. [Google Scholar]
- Steele, P.R.; Hertweck, K.L.; Mayfield, D.; McKain, M.R.; Leebens-Mack, J.; Pires, J.C. Quality and quantity of data recovered from massively parallel sequencing: Examples in Asparagales and Poaceae. Am. J. Bot. 2012, 99, 330–348. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Dou, S.; Ji, Z.; Xue, Q. Synonymous codon usage and gene function are strongly related in Oryza sativa. Biosystems 2005, 80, 123–131. [Google Scholar] [CrossRef]
- Campbell, W.H.; Gowri, G. Codon usage in higher plants, green algae, and cyanobacteria. Plant Physiol. 1990, 92, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, D.; Shanker, A. Identification of simple sequence repeats in chloroplast genomes of Magnoliids through bioinformatics approach. Interdiscip. Sci. Comput. Life Sci. 2016, 8, 327–336. [Google Scholar] [CrossRef]
- Li, Y.; Kuang, X.J.; Zhu, X.X.; Zhu, Y.; Sun, C. Codon usage bias of Catharanthus roseus. Zhongguo Zhong Yao Za Zhi = Zhongguo Zhongyao Zazhi = China J. Chin. Mater. Med. 2016, 41, 4165–4168. [Google Scholar]
- Wu, X.-M.; Wu, S.-F.; Ren, D.-M.; Zhu, Y.-P.; He, F.-C. The analysis method and progress in the study of codon bias. Yi Chuan = Hered. 2007, 29, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.M.; Gladyshev, V.N. Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 2012, 287, 4419–4425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Li, W.-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Xue, Q. Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 2005, 84, 55–62. [Google Scholar] [CrossRef]
- Park, I.; Kim, W.; Yeo, S.-M.; Choi, G.; Kang, Y.-M.; Piao, R.; Moon, B. The complete chloroplast genome sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef] [Green Version]
- Zuo, L.-H.; Shang, A.-Q.; Zhang, S.; Yu, X.-Y.; Ren, Y.-C.; Yang, M.-S.; Wang, J.-M. The first complete chloroplast genome sequences of Ulmus species by de novo sequencing: Genome comparative and taxonomic position analysis. PLoS ONE 2017, 12, e0171264. [Google Scholar] [CrossRef] [Green Version]
- Freyer, R.; Hoch, B.; Neckermann, K.; Maier, R.M.; Kössel, H. RNA editing in maize chloroplasts is a processing step independent of splicing and cleavage to monocistronic mRNAs. Plant J. 1993, 4, 621–629. [Google Scholar] [CrossRef]
- Tang, W.; Luo, C. Molecular and functional diversity of RNA editing in plant mitochondria. Mol. Biotechnol. 2018, 60, 935–945. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Corneille, S.; Lutz, K.; Maliga, P. Conservation of RNA editing between rice and maize plastids: Are most editing events dispensable? Mol. Gen. Genet. 2000, 264, 419–424. [Google Scholar] [CrossRef]
- Jain, B.P.; Chauhan, P.; Tanti, G.K.; Singarapu, N.; Ghaskadbi, S.; Goswami, S.K. Tissue specific expression of SG2NA is regulated by differential splicing, RNA editing and differential polyadenylation. Gene 2015, 556, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.; Coates, D. The code within the codons. Biosystems 1989, 22, 177–187. [Google Scholar] [CrossRef]
- Qi, W.; Lin, F.; Liu, Y.; Huang, B.; Cheng, J.; Zhang, W.; Zhao, H. High-throughput development of simple sequence repeat markers for genetic diversity research in Crambe abyssinica. BMC Plant Biol. 2016, 16, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, D.-Y.; Wu, H.; Wang, Y.-L.; Gao, L.-M.; Zhang, S.-Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Chen, X.; Cui, Y.; Sun, W.; Li, Y.; Wang, Y.; Song, J.; Yao, H. Molecular structure and phylogenetic analyses of complete chloroplast genomes of two Aristolochia medicinal species. Int. J. Mol. Sci. 2017, 18, 1839. [Google Scholar] [CrossRef]
- Qi, W.-H.; Jiang, X.-M.; Yan, C.-C.; Zhang, W.-Q.; Xiao, G.-S.; Yue, B.-S.; Zhou, C.-Q. Distribution patterns and variation analysis of simple sequence repeats in different genomic regions of bovid genomes. Sci. Rep. 2018, 8, 14407. [Google Scholar] [CrossRef]
- Wang, W.; Messing, J. High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hall, N.; McElroy, J.S.; Lowe, E.K.; Goertzen, L.R. Complete plastid genome sequence of goosegrass (Eleusine indica) and comparison with other Poaceae. Gene 2017, 600, 36–43. [Google Scholar] [CrossRef]
- Yang, J.-B.; Tang, M.; Li, H.-T.; Zhang, Z.-R.; Li, D.-Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [Green Version]
- Strauss, S.H.; Palmer, J.D.; Howe, G.T.; Doerksen, A.H. Chloroplast genomes of two conifers lack a large inverted repeat and are extensively rearranged. Proc. Natl. Acad. Sci. USA 1988, 85, 3898–3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Cui, L.; Feng, K.; Deng, P.; Du, X.; Wan, F.; Weining, S.; Nie, X. Comparative analysis of Asteraceae chloroplast genomes: Structural organization, RNA editing and evolution. Plant Mol. Biol. Report. 2015, 33, 1526–1538. [Google Scholar] [CrossRef]
- Yu, X.; Tan, W.; Zhang, H.; Gao, H.; Wang, W.; Tian, X. Complete Chloroplast Genomes of Ampelopsis humulifolia and Ampelopsis japonica: Molecular Structure, Comparative Analysis, and Phylogenetic Analysis. Plants 2019, 8, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, U.J.B.; Nunes, R.; Targueta, C.P.; Diniz-Filho, J.A.F.; de Campos Telles, M.P. The complete chloroplast genome of Stryphnodendron adstringens (Leguminosae-Caesalpinioideae): Comparative analysis with related Mimosoid species. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef]
- Ma, P.-F.; Zhang, Y.-X.; Zeng, C.-X.; Guo, Z.-H.; Li, D.-Z. Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 2014, 63, 933–950. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Yu, M.; Wang, X.; Zhang, X.-H. Comparative genomic analysis reveals the evolution and environmental adaptation strategies of vibrios. BMC Genom. 2018, 19, 135. [Google Scholar] [CrossRef] [Green Version]
- Tonti-Filippini, J.; Nevill, P.G.; Dixon, K.; Small, I. What can we do with 1000 plastid genomes? Plant J. 2017, 90, 808–818. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Kunth, C. Melanthaceae [sic]. Enumeratio Plant. 1843, 4, 1. [Google Scholar]
- Wang, Y.; Sun, J.; Yuan, Q.; Guo, L. Complete chloroplast genome sequence of Dracaena cochinchinensis: Genome structure and genomic resources. Mitochondrial DNA Part B 2019, 4, 1750–1751. [Google Scholar] [CrossRef] [Green Version]
- Pfanzelt, S.; Albach, D.C.; von Hagen, K.B. Extremely low levels of chloroplast genome sequence variability in Astelia pumila (Asteliaceae, Asparagales). PeerJ 2019, 7, e6244. [Google Scholar] [CrossRef] [Green Version]
- Chase, M.W.; Hills, H.H. Silica gel: An ideal material for field preservation of leaf samples for DNA studies. Taxon 1991, 40, 215–220. [Google Scholar] [CrossRef]
- Doyle, J. DNA protocols for plants. In Molecular Techniques in Taxonomy; Hewitt, G.M., Johnston, A.W.B., Young, J.P.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 283–293. [Google Scholar]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A simple and fast pipeline for de novo assembly of a complete circular chloroplast genome using genome skimming data. BioRxiv 2018, 256479. [Google Scholar] [CrossRef] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M. Phylogeny. fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Chevenet, F.; Brun, C.; Bañuls, A.-L.; Jacq, B.; Christen, R. TreeDyn: Towards dynamic graphics and annotations for analyses of trees. BMC Bioinform. 2006, 7, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | Chlorophytum comosum | Chlorophytum gallabatense | Allium victorialis | Asparagus officinalis | Anemarrhena asphodeloides | Anthericum ramosum | Chlorophytum rhizopendulum |
|---|---|---|---|---|---|---|---|
| Size (bp) | 154,248 | 154,154 | 154,074 | 156,699 | 156,917 | 155,812 | 153,504 |
| LSC (bp) | 84,004 | 83,686 | 83,169 | 84,999 | 85,027 | 84,817 | 93,446 |
| IR (bp) | 52,228 | 52,508 | 53,052 | 53,062 | 53,320 | 52,756 | 42,102 |
| SSC (bp) | 18,016 | 17,960 | 17,853 | 18,638 | 18,570 | 18,239 | 17,956 |
| Total genes | 112 | 112 | 116 | 112 | 112 | 114 | 112 |
| PCGs | 78 | 78 | 82 | 78 | 78 | 80 | 78 |
| tRNA genes | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
| rRNA genes | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Adenine (A) | 31.0% | 31.0% | 31.2% | 30.9% | 30.8% | 31.0% | 31.0% |
| Thymine (T) | 31.7% | 31.7% | 31.8% | 31.5% | 31.4% | 31.7% | 31.7% |
| Guanine (G) | 18.3% | 18.3% | 18.2% | 18.5% | 18.6% | 18.3% | 18.3% |
| Cytosine (C) | 19.0% | 19.0% | 18.8% | 19.1% | 19.3% | 19.0% | 19.0% |
| GC content | 37.3% | 37.3% | 37.0% | 37.6% | 37.8% | 37.3% | 37.3% |
| Category | Group of Genes | Name of Genes |
|---|---|---|
| Self-replication | Large subunit ribosomal proteins | rpl2a,b, rpl14, rpl16, rpl20, rpl22, rpl23a, rpl32, rpl33, rpl36 |
| Small subunit ribosomal proteins | ψrps2, rps3, rps4, rps7a, rps8, rps11, rps12a,b, rps14, rps15, rps16b, rps18 | |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1b, rpoC2 | |
| rRNA genes | rrn4.5, rrn5, rrn16, rrn23 | |
| tRNA genes | trnl-CAUa, trnL-CAAa, trnV-GACa, trnl-GAUa,b, trnA-UGCa,b, trnR-ACGa, trnN-GUUa, trnL-UAG, trnP-UGG, trnW-CCA, trnQ-UUG, trnS-GCU, trnR-UCU, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnT-GGU, trnS-UGA, trnG-UCCb, trnG-GCC, trnfM-CAU, trnS-GGA, trnH-GUGa, trnT-UGU, trnL-UAAb, trnF-GAA, trnV-UACb, trnM-CAU, trnK-UUUb | |
| photosynthesis | Photosystem I | psaA, psaB, psaC, psal, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbl, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Cytochrome b/f complex | petA, petB, petD, petG, petL, petN | |
| ATP synthase | atpA, atpB, atpE, atpFb, atpH, atpl | |
| RuBisCO | rbcL | |
| Subunits of NADH-dehydrogenase | ndhAb, ndhBa,b, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhl, ndhJ, ndhK | |
| Other genes | Maturase | MatK |
| Proteolysis | clpPc | |
| Translation initiation factor | ψinfA | |
| Carbon metabolism | cemA | |
| Fatty acid synthesis | accD | |
| Cytochrome c synthesis gene | ccsA | |
| unknown | Conserved open reading frames | Ycf1a, ycf2a, ycf3c, ycf4 |
| SSR Type | Repeat Unit | Amount | Ratio (%) | ||
|---|---|---|---|---|---|
| C. comosum | C. gallabatense | C. comosum | C. gallabatense | ||
| Mono | A/T | 86 | 68 | 100% | 100% |
| Di | AG/CT | 3 | 3 | 21.4% | 25% |
| AT/AT | 11 | 9 | 78.6% | 75% | |
| Tri | AAT/ATT | 2 | 1 | 100% | 100% |
| Tetra | AAAG/CTTT | 1 | 1 | 11.1% | 12.5% |
| AAAT/ATTT | 6 | 6 | 66.7% | 75% | |
| AATG/ATTC | 1 | 0 | 11.1% | 0% | |
| ACAT/ATGT | 1 | 1 | 11.1% | 12.5% | |
| Penta | AAGAT/ATCTT | 0 | 1 | 0% | 100% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munyao, J.N.; Dong, X.; Yang, J.-X.; Mbandi, E.M.; Wanga, V.O.; Oulo, M.A.; Saina, J.K.; Musili, P.M.; Hu, G.-W. Complete Chloroplast Genomes of Chlorophytum comosum and Chlorophytum gallabatense: Genome Structures, Comparative and Phylogenetic Analysis. Plants 2020, 9, 296. https://doi.org/10.3390/plants9030296
Munyao JN, Dong X, Yang J-X, Mbandi EM, Wanga VO, Oulo MA, Saina JK, Musili PM, Hu G-W. Complete Chloroplast Genomes of Chlorophytum comosum and Chlorophytum gallabatense: Genome Structures, Comparative and Phylogenetic Analysis. Plants. 2020; 9(3):296. https://doi.org/10.3390/plants9030296
Chicago/Turabian StyleMunyao, Jacinta N., Xiang Dong, Jia-Xin Yang, Elijah M. Mbandi, Vincent O. Wanga, Millicent A. Oulo, Josphat K. Saina, Paul M. Musili, and Guang-Wan Hu. 2020. "Complete Chloroplast Genomes of Chlorophytum comosum and Chlorophytum gallabatense: Genome Structures, Comparative and Phylogenetic Analysis" Plants 9, no. 3: 296. https://doi.org/10.3390/plants9030296