Comparative Genomics and Phylogenetic Analyses of Christia vespertilionis and Urariopsis brevissima in the Tribe Desmodieae (Fabaceae: Papilionoideae) Based on Complete Chloroplast Genomes


Abstract
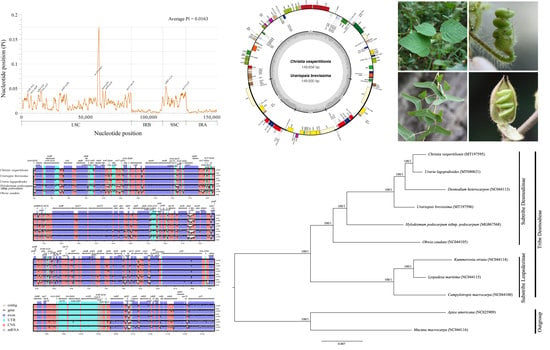
:
1. Introduction
2. Results
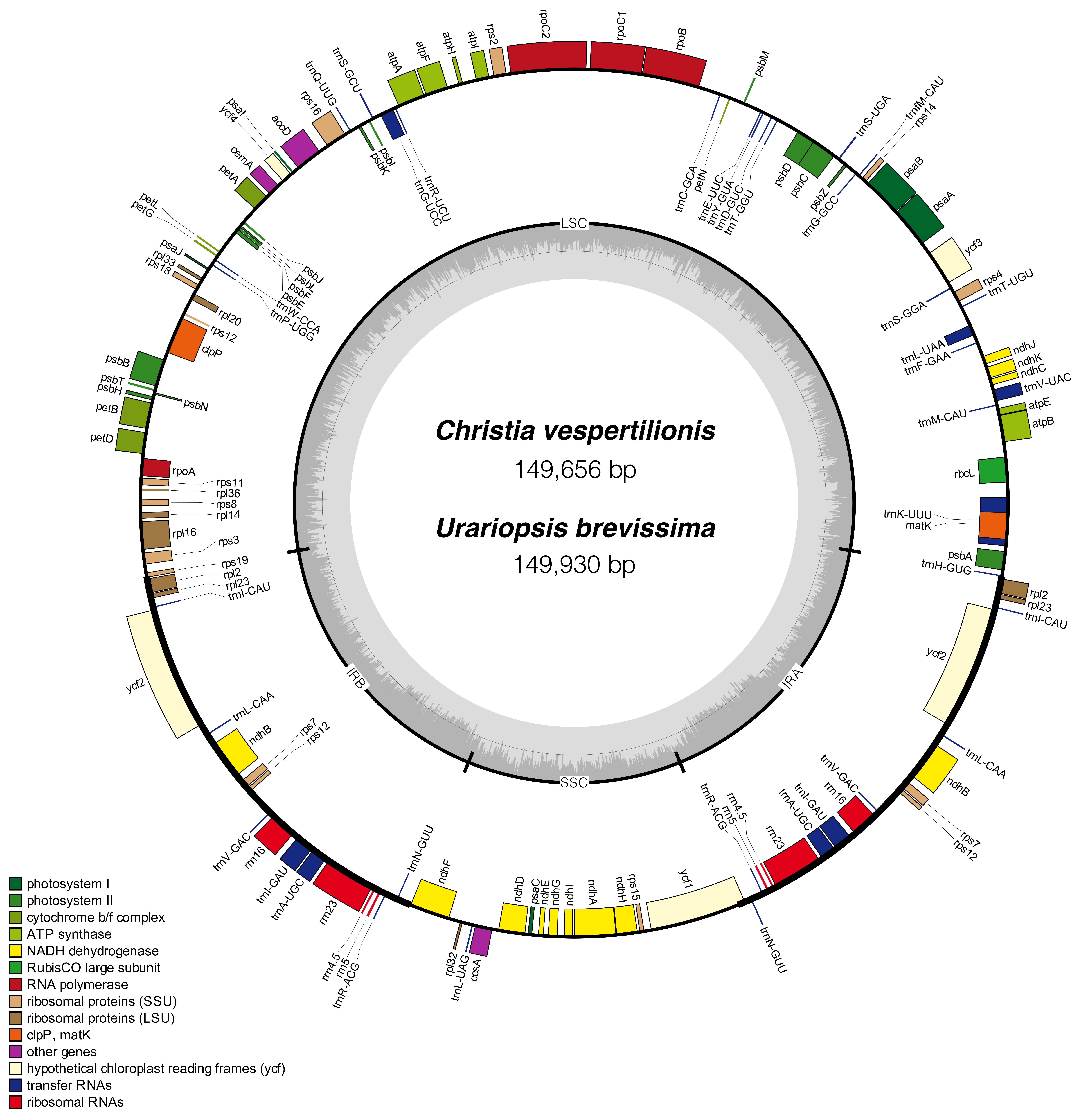
2.1. Genome Structural Features and Gene Content
2.2. Amino Acid Abundance and Codon Usage
2.3. Simple Sequence Repeats and Repetitive Sequence Analysis
2.4. Sequence Divergence Analysis
2.5. Phylogenetic Analysis
3. Discussion
3.1. Comparative Analysis of Desmodieae Chloroplast Genomes
3.2. Phylogenetic Relationships of Desmodieae
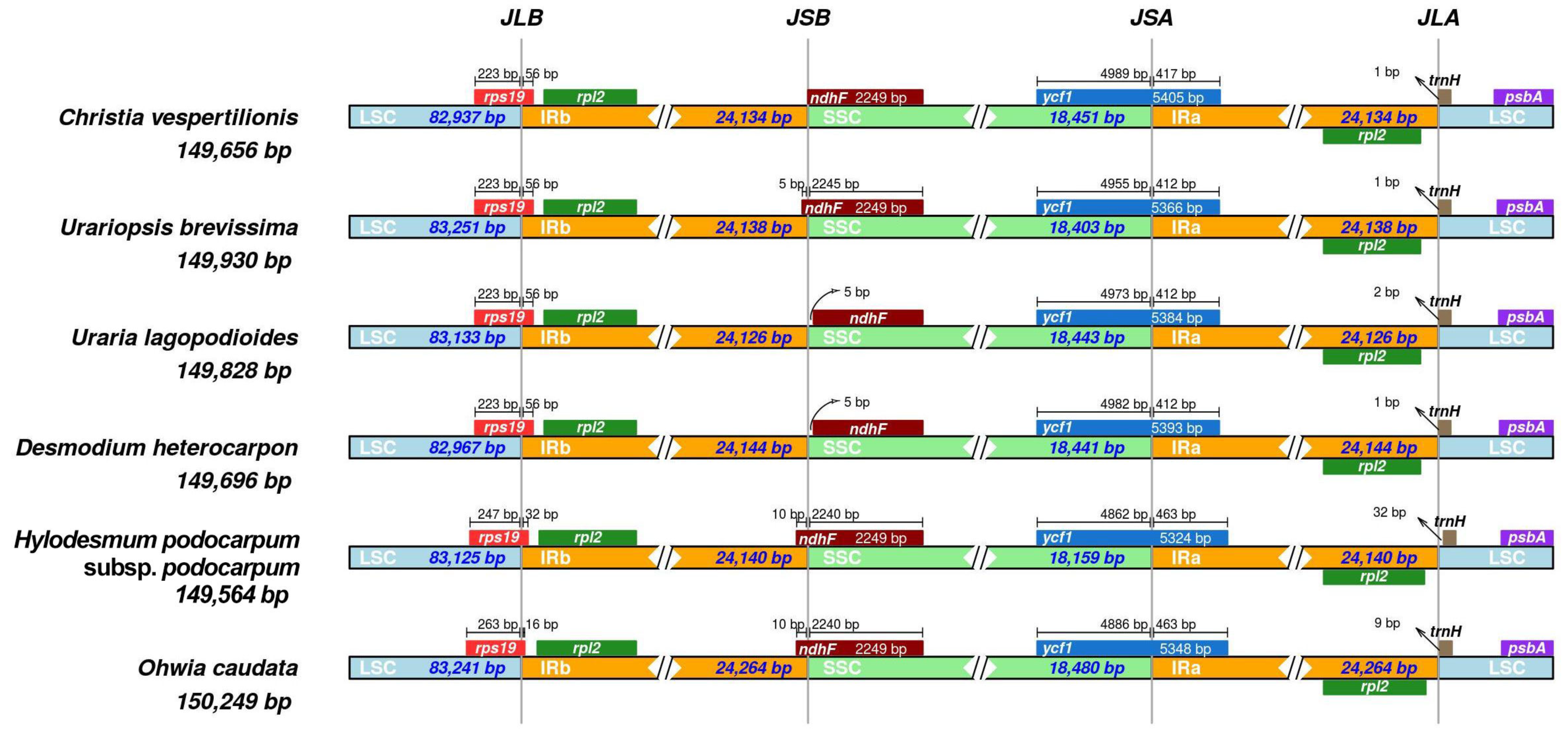
3.3. Identification of Highly Variable Regions
4. Materials and Methods
4.1. Material Sampling and DNA Extraction
4.2. Genome Sequencing, Assembly, and Annotation
4.3. Codon Usage
4.4. Simple Sequence Repeats and Repetitive Sequence Analysis
4.5. Genome Comparison and Divergent Hotspots Identification
4.6. Phylogeneitc Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lewis, G.P. Legumes of the World; Royal Botanic Gardens Kew: Richmond, UK, 2005; p. 592. [Google Scholar]
- Bruneau, A.; Doyle, J.J.; Herendeen, P.; Hughes, C.; Kenicer, G.; Lewis, G.; Mackinder, B.; Pennington, R.T.; Sanderson, M.J.; Wojciechowski, M.F.; et al. Legume phylogeny and classification in the 21st century: Progress, prospects and lessons for other species-rich clades. Taxon 2013, 62, 217–248. [Google Scholar] [CrossRef]
- Azani, N.; Babineau, M.; Bailey, C.D.; Banks, H.; Barbosa, A.R.; Pinto, R.B.; Boatwright, J.S.; Borges, L.M.; Brown, G.K.; Bruneau, A.; et al. A new subfamily classification of the Leguminosae based on a taxonomically comprehensive phylogeny. Taxon 2017, 66, 44–77. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, H. Desmodieae. In Legumes of the World; Lewis, G.P., Ed.; Royal Botanic Gardens Kew: Richmond, UK, 2005; pp. 433–446. [Google Scholar]
- Jabbour, F.; Gaudeul, M.; Lambourdiere, J.; Ramstein, G.; Hassanin, A.; Labat, J.N.; Sarthou, C. Phylogeny, biogeography and character evolution in the tribe Desmodieae (Fabaceae: Papilionoideae), with special emphasis on the New Caledonian endemic genera. Mol. Phylogenet. Evol. 2018, 118, 108–121. [Google Scholar] [CrossRef]
- Huang, P.H.; Ohashi, H.; Oikawa, Y. Uraria Desvaux. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2010; Volume 10, pp. 286–288. [Google Scholar]
- Dehaas, A.J.P.; Bosman, M.T.M.; Geesink, R. Urariopsis reduced to Uraria (Leguminosae-Papilionoideae). Blumea 1980, 26, 439–444. [Google Scholar]
- Ohashi, H.; Iokawa, Y.; Phon, P.D. The genus Uraria (Leguminosae) in China. J. Jpn. Bot. 2006, 81, 332–361. [Google Scholar]
- Ohashi, K.; Ohashi, H.; Nemoto, T.; Ikeda, T.; Izumi, H.; Kobayashi, H.; Muragaki, H.; Nata, K.; Sato, N.; Suzuki, M. Phylogenetic analyses for a new classification of the Desmodium Group of Leguminosae tribe Desmodieae. Shokubutsu Kenkyu Zasshi 2018, 93, 165–189. [Google Scholar]
- Neuhaus, H.E.; Emes, M.J. Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Biol. 2000, 51, 111–140. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.R.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Ser. B Biol. Sci. 2003, 358, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Wicke, S.; Schneeweiss, G.M.; de Pamphilis, C.W.; Muller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.P.; Choi, I.S.; Choi, B.H. Plastid genome evolution in tribe Desmodieae (Fabaceae: Papilionoideae). PLoS ONE 2019, 14, e0218743. [Google Scholar] [CrossRef] [Green Version]
- Somaratne, Y.; Guan, D.L.; Wang, W.Q.; Zhao, L.; Xu, S.Q. The complete chloroplast genomes of two Lespedeza species: Insights into codon usage bias, RNA editing sites, and phylogenetic relationships in Desmodieae (Fabaceae: Papilionoideae). Plants (Basel) 2019, 9, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2016, 7, 2054. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wu, X.; Zhang, D. Phylogenetic analysis of Fritillaria cirrhosa D. Don and its closely related species based on complete chloroplast genomes. PeerJ 2019, 7, e7480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Ge, X.; Cano, A.; Salazar, B.G.M.; Deng, Y. Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): Molecular structure, phylogenetic relationships, and adaptive evolution. PeerJ 2020, 8, e8450. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.H.; Wicke, S.; Wang, H.; Jin, J.J.; Chen, S.Y.; Zhang, S.D.; Li, D.Z.; Yi, T.S. Plastid genome evolution in the early-diverging legume subfamily Cercidoideae (Fabaceae). Front. Plant Sci. 2018, 9, 138. [Google Scholar] [CrossRef] [Green Version]
- Koenen, E.J.; Ojeda, D.I.; Steeves, R.; Migliore, J.; Bakker, F.T.; Wieringa, J.J.; Kidner, C.; Hardy, O.J.; Pennington, R.T.; Bruneau, A. Large-scale genomic sequence data resolve the deepest divergences in the legume phylogeny and support a near-simultaneous evolutionary origin of all six subfamilies. New Phytol. 2020, 225, 1355–1369. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Wang, Y.-H.; Jin, J.-J.; Stull, G.W.; Bruneau, A.; Cardoso, D.; de Queiroz, L.P.; Moore, M.J.; Zhang, S.-D.; Chen, S.-Y. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst. Biol. 2020, in press. [Google Scholar] [CrossRef]
- Zhao, X.L.; Zhu, Z.M. The complete chloroplast genome of Uraria lagopodioides (Fabaceae). Mitochondrial DNA B 2020, 5, 1365–1366. [Google Scholar] [CrossRef] [Green Version]
- Dubchak, I.; Ryaboy, D.V. VISTA family of computational tools for comparative analysis of DNA sequences and whole genomes. In Gene Mapping, Discovery, and Expression; Humana Press: Totowa, NJ, USA, 2006; pp. 69–89. [Google Scholar]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef]
- Leebens-Mack, J.; Raubeson, L.A.; Cui, L.Y.; Kuehl, J.V.; Fourcade, M.H.; Chumley, T.W.; Boore, J.L.; Jansen, R.K.; de Pamphilis, C.W. Identifying the basal angiosperm node in chloroplast genome phylogenies: Sampling one’s way out of the felsenstein zone. Mol. Biol. Evol. 2005, 22, 1948–1963. [Google Scholar] [CrossRef]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S.M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.H.; Li, S.; Qian, Z.Q.; Su, H.L.; Huang, Z.H.; Guo, Z.G.; Dai, P.F.; Liu, Z.L.; Zhao, G.F. Strong phylogeographic pattern of cpDNA variation reveals multiple glacial refugia for Saruma henryi Oliv. (Aristolochiaceae), an endangered herb endemic to China. Mol. Phylogenet Evol. 2010, 57, 176–188. [Google Scholar] [CrossRef]
- He, S.; Wang, Y.; Volis, S.; Li, D.; Yi, T. Genetic diversity and population structure: Implications for conservation of wild soybean (Glycine soja Sieb. et Zucc) based on nuclear and chloroplast microsatellite variation. Int. J. Mol. Sci. 2012, 13, 12608–12628. [Google Scholar] [CrossRef] [Green Version]
- Perdereau, A.C.; Kelleher, C.T.; Douglas, G.C.; Hodkinson, T.R. High levels of gene flow and genetic diversity in Irish populations of Salix caprea L. inferred from chloroplast and nuclear SSR markers. BMC Plant Biol. 2014, 14, 202. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.K.; Kaittanis, C.; Saski, C.; Lee, S.B.; Tomkins, J.; Alverson, A.J.; Daniell, H. Phylogenetic analyses of Vitis (Vitaceae) based on complete chloroplast genome sequences: Effects of taxon sampling and phylogenetic methods on resolving relationships among rosids. BMC Evol. Biol. 2006, 6, 32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Du, L.; Liu, A.; Chen, J.; Wu, L.; Hu, W.; Zhang, W.; Kim, K.; Lee, S.C.; Yang, T.J.; et al. The Complete chloroplast genome sequences of five Epimedium species: Lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 2016, 7, 306. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Ohashi, H. Morphology and polymorphism in the pollen of Christia and Uraria (Leguminosae: Papillionoideae). J. Jpn. Bot. 2002, 77, 150–162. [Google Scholar]
- Schindler, A.K. Desmodiinae novae. Englers Bot. Jahrbücher 1916, 54, 51–53. [Google Scholar]
- Ohashi, H. The Asiatic species of Desmodium and its allied genera (Leguminosae). Ginkgoana 1973, 1, 1–318. [Google Scholar]
- Gagnepain, F. Uraria Desv. In Flore Générale de L’indochine; Lecomte, M.H., Ed.; Masson: Paris, France, 1920; Volume 2, pp. 538–551. [Google Scholar]
- Van Meeuwen, M.S.; Nooteboom, H.P.; van Steenis, C.G.G.J. Preliminary revisions of 563 some genera of Malaysian Papilionaceae I. Reinwardtia 1961, 5, 419–456. [Google Scholar]
- Yang, Y.C.; Huang, P.H. A revision of the genus Uraria Desv. (Leguminosae) in China. Bull. Bot. Res. 1981, 1, 1–20. [Google Scholar]
- Hutchinson, J. The Genera of Flowering Plants; Clarendon Press: Oxford, UK, 1964; Volume 1, p. 516. [Google Scholar]
- Duminil, J.; Heuertz, M.; Doucet, J.L.; Bourland, N.; Cruaud, C.; Gavory, F.; Doumenge, C.; Navascués, M.; Hardy, O.J. CpDNA-based species identification and phylogeography: Application to African tropical tree species. Mol. Ecol. 2010, 19, 5469–5483. [Google Scholar] [CrossRef]
- Wambulwa, M.C.; Meegahakumbura, M.K.; Kamunya, S.; Muchugi, A.; Moller, M.; Liu, J.; Xu, J.C.; Ranjitkar, S.; Li, D.Z.; Gao, L.M. Insights into the genetic relationships and breeding patterns of the African tea germplasm based on nSSR markers and cpDNA sequences. Front. Plant Sci. 2016, 7, 1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Tan, W.; Sun, J.; Du, J.; Zheng, C.; Tian, X.; Zheng, M.; Xiang, B.; Wang, Y. Comparison of four complete chloroplast genomes of medicinal and ornamental Meconopsis species: Genome organization and species discrimination. Sci. Rep. 2019, 9, 10567. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Lou, G.; Cai, X.; Zhang, B.; Cheng, Y.; Wang, H. Comparison of the complete plastomes and the phylogenetic analysis of Paulownia species. Sci. Rep. 2020, 10, 2225. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, X.J.; Moore, M.J.; Li, D.Z.; Yi, T.S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA genes in genomic sequences. In Gene Prediction; Humana Press: New York, NY, USA, 2019; pp. 1–14. [Google Scholar]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Features | Christia vespertilionis | Urariopsis brevissima | Uraria lagopodioides | Desmodium heterocarpon | Hylodesmum podocarpum subsp. podocarpum | Ohwia caudata |
|---|---|---|---|---|---|---|
| Accession number | MT197595 | MT197596 | MT040621 | NC044113 | MG867568 | NC044105 |
| Genome size (bp) | 149,656 | 149,930 | 149,828 | 149,696 | 149,564 | 150,249 |
| LSC length (bp) | 82,937 | 83,251 | 83,133 | 82,967 | 83,125 | 83,241 |
| SSC length (bp) | 18,451 | 18,403 | 18,443 | 18,441 | 18,159 | 18,480 |
| IR length (bp) | 24,134 | 24,138 | 24,126 | 24,144 | 24,140 | 24,264 |
| PCGs total length (bp) | 78,063 | 78,084 | 78,030 | 77,427 | 77,925 | 78,045 |
| Total number of genes | 128 | 128 | 128 | 128 | 128 | 128 |
| Protein-coding genes | 83 | 83 | 83 | 83 | 83 | 83 |
| tRNA | 37 | 37 | 37 | 37 | 37 | 37 |
| rRNA | 8 | 8 | 8 | 8 | 8 | 8 |
| Overall GC% | 35.2 | 35.2 | 35.2 | 35.2 | 35.2 | 35.1 |
| GC% in LSC | 32.8 | 32.7 | 32.7 | 32.7 | 32.6 | 32.6 |
| GC% in SSC | 28.2 | 28.2 | 28.1 | 28.1 | 28.5 | 28.3 |
| GC% in IR | 42.1 | 42.1 | 42.1 | 42.1 | 42.1 | 42 |
| GC% in PCGs | 36.2 | 36.1 | 36.2 | 36.1 | 36.1 | 36 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, X.-L.; Zhu, Z.-M. Comparative Genomics and Phylogenetic Analyses of Christia vespertilionis and Urariopsis brevissima in the Tribe Desmodieae (Fabaceae: Papilionoideae) Based on Complete Chloroplast Genomes. Plants 2020, 9, 1116. https://doi.org/10.3390/plants9091116
Zhao X-L, Zhu Z-M. Comparative Genomics and Phylogenetic Analyses of Christia vespertilionis and Urariopsis brevissima in the Tribe Desmodieae (Fabaceae: Papilionoideae) Based on Complete Chloroplast Genomes. Plants. 2020; 9(9):1116. https://doi.org/10.3390/plants9091116
Chicago/Turabian StyleZhao, Xue-Li, and Zhang-Ming Zhu. 2020. "Comparative Genomics and Phylogenetic Analyses of Christia vespertilionis and Urariopsis brevissima in the Tribe Desmodieae (Fabaceae: Papilionoideae) Based on Complete Chloroplast Genomes" Plants 9, no. 9: 1116. https://doi.org/10.3390/plants9091116