
Using Genealogical Concordance and Coalescent-Based Species Delimitation to Assess Species Boundaries in the Diaporthe eres Complex



Centre for Environmental and Marine Studies (CESAM), Department of Biology, Campus Universitário de Santiago, University of Aveiro, 3810-193 Aveiro, Portugal
*
Author to whom correspondence should be addressed.
J. Fungi 2021, 7(7), 507; https://doi.org/10.3390/jof7070507
Submission received: 24 May 2021
/
Revised: 11 June 2021
/
Accepted: 23 June 2021
/
Published: 25 June 2021
(This article belongs to the Section Fungal Evolution, Biodiversity and Systematics)
Abstract
:DNA sequence analysis has been of the utmost importance to delimit species boundaries in the genus Diaporthe. However, the common practice of combining multiple genes, without applying the genealogical concordance criterion has complicated the robust delimitation of species, given that phylogenetic incongruence between loci has been disregarded. Despite the several attempts to delineate the species boundaries in the D. eres complex, the phylogenetic limits within this complex remain unclear. In order to bridge this gap, we employed the Genealogical Phylogenetic Species Recognition principle (GCPSR) and the coalescent-based model Poisson Tree Processes (PTPs) and evaluated the presence of recombination within the D. eres complex. Based on the GCPSR principle, presence of incongruence between individual gene genealogies, i.e., conflicting nodes and branches lacking phylogenetic support, was evident. Moreover, the results of the coalescent model identified D. eres complex as a single species, which was not consistent with the current large number of species within the complex recognized in phylogenetic analyses. The absence of reproductive isolation and barriers to gene flow as well as the high haplotype and low nucleotide diversity indices within the above-mentioned complex suggest that D. eres constitutes a population rather than different lineages. Therefore, we argue that a cohesive approach comprising genealogical concordance criteria and methods to detect recombination must be implemented in future studies to circumscribe species in the genus Diaporthe.
1. Introduction
A reliable and accurate identification of fungal plant pathogens is of the utmost importance in disease diagnosis to implement effective management and quarantine strategies [1]. However, one crucial aspect in recognizing a fungal species is correctly defining a species [2]. Although molecular approaches have changed our perception of fungal diversity [3], difficulties in understanding the evolutionary processes in fungi have arisen, thus turning a correct definition of a fungal species into a prevailing challenge to mycologists [4,5].
The Genealogical Concordance Phylogenetic Species Recognition (GCPSR), introduced by Taylor et al. [6], relies in the comparison of individual gene genealogies to identify incongruences, and it has been particularly useful to delimit the species boundaries in morphologically conserved fungi [7]. However, the common approach of concatenating sequence data to delimit species without following the GCPSR principle [6,8] overestimates the true diversity of species, since each clade in combined trees is frequently recognized as a distinct lineage [9,10]. Moreover, species boundaries in closely related taxa can be somehow difficult to determine through multilocus sequence data, as some alleles are not expected to be reciprocally monophyletic in the initial stages of speciation, resulting in phylogenetic incongruences [7,11]. As an alternative, the delimitation of species using multi-species coalescent models provides a more comprehensive insight into the speciation events, as it can estimate species boundaries even in the presence of incongruence between individual genealogies [12].
Some authors have already advocated that the description of fungal species needs to be based on approaches that employ a combination of several methods: phylogenetic analysis following the genealogical concordance (GCPSR) [6], coalescence models such as Poisson Tree Processes (PTPs) [13], and splits graphs (Phylogenetic networks) [2]. Nevertheless, despite the usefulness of the above-mentioned methods to support the boundaries of species, there are only a few studies on fungi, namely in Colletotrichum [7], Beauveria [14] and more recently, Diaporthe [15].
Diaporthe species are associated with a wide range of plant hosts as pathogens, endophytes or saprobes of crops, ornamentals, and forest trees [16,17,18]. Species identification in the genus Diaporthe has evolved from host association and morphology [19,20] to the widespread adoption of DNA sequencing [21]. Currently, the nuclear ribosomal internal transcribed spacer (ITS), the translation elongation factor 1-α (TEF1-α), beta-tubulin (TUB2), histone H3 (HIS3), and calmodulin (CAL) genes are the most used molecular loci in this genus, which currently includes the description of over 200 species supported by ex-type cultures and supplementary DNA sequences [4,22].
Diaporthe eres is the type species of the genus and was originally described by Nitschke (1870), from Ulmus sp. in Germany [23]. In 1933, Wehmeyer listed a number of synonyms under D. eres based on morphological characters. Later, based on molecular data, few of these synonyms were accepted by several authors [21,23]. Udayanga et al. [23], attempted to define the species limits of D. eres and closely related species based on the concatenation of seven loci and designated an epitype specimen for D. eres. Despite the attempts by several authors to re-examine the D. eres complex, its boundaries are not entirely understood [17,18,24]. In several studies, many species have been demonstrated to be synonymous to the D. eres species, based on multi-locus analyses such us D. biguttusis, D. camptothecicola, D. castaneae-mollissimae, D. cotoneastri, D. ellipicola, D. longicicola, D. mahothocarpus, D. momicola and Phomopsis fukushii [18,23,24]. In a recent study, Chaisiri et al. [25] found that D. henanensis, D. lonicerae and D. rosicola were also synonyms of D. eres, based on the GCPSR principle coupled with haplotype network analysis and population genetic diversity. Although D. eres has been regarded as a minor pathogen, it is also considered one of the main causal agents of cankers on grapevines [17], blueberries [26] and apple trees [27]. Lopes et al. [28] reported the occurrence of D. eres in forest trees but found some difficulties in the interpretation of their phylogenetic analyses and delimitation of the species in the D. eres complex. Therefore, the aim of this study was to clarify the limits of the D. eres species complex by implementing several distinct methods, such as single and multilocus phylogenetic analyses, the genealogical concordance principle, coalescent-based species delimitation methods (PTP), phylogenetic networks, pairwise homoplasy index test and comparison of morphological characters.
2. Materials and Methods
2.1. Fungal Isolates
In 2007, a survey was conducted to inspect for the presence of Diaporthe species associated with ornamental plants in Portugal. Twigs and leaves of Banksia sp. showing blight symptoms typical of Diaporthe were collected. Fungal isolates were obtained by the methods described by Hilário et al. [29], from which a fungus was collected that resembled D. eres. Moreover, isolates CAA954 and CAA1001 obtained previously from Quercus suber and Pinus pinaster and identified as D. eres [28] were also included in this study. Cultures were maintained in the collection of Artur Alves (CAA), University of Aveiro (Portugal), on potato dextrose agar (PDA) (Merck, Darmstadt, DE, Germany). The isolates used in this study are listed in Table 1.
2.2. DNA Extraction and PCR Amplification
The genomic DNA was extracted from 7-day-old fungal isolates, grown at 25 °C, according to a modified protocol of Möller [30]. The nuclear ribosomal internal transcribed spacer region using the primers set ITS5 and NL4 [31] was amplified as described by Alves et al. [32]. The primers set EF-688F/EF-1251R [33], T1/Bt2b [34,35], CYLH3F/H3-1bR [34,36] and CAL-38F/CAL-737R [29,37] were used to amplify part of TEF1-α, TUB2, HIS3 and CAL loci, respectively.
All PCR reaction mixtures, with a final volume of 25 µL, were composed of 15.75 µL of sterile pure water, 6.25 µL of NZYTaq 2xgreen Master Mix (NzytechTM, Lisbon, PT, Portugal), 1 µL of each primer at 10 pmol/µL and 1 µL of DNA template. PCR reactions were performed as described by Hilário et al. [28]. The amplified PCR fragments were purified using the NZYGelPure Kit (NzytechTM, Lisbon, Portugal) and sequenced by GATC Biotech (Cologne, DE, Germany).
2.3. Phylogenetic Analyses
Multilocus phylogenetic analyses based on 5 loci (ITS, TEF1-α, TUB2, HIS3 and CAL) and 4 loci (TEF1-α, TUB2, HIS3 and CAL) were performed for the Diaporthe eres species complex. The alignments of different gene regions, including sequences of the isolates used in this study and those retrieved from GenBank (Table 1), were performed with ClustalX2.1 software [38] using the following parameters: pairwise alignment (gap opening = 10, gap extension = 0.1) and multiple alignment (gap opening = 10, gap extension = 0.2, transition weight = 0.5, delay divergent sequences = 25%). The alignments were checked and edited manually with BioEdit Alignment Editor v.7.0.5.3 [39] and concatenated using Sequence Matrix software [40]. Phylogenetic analyses of sequence data were done using PAUP* v.4.0b10 [41] for Maximum Parsimony (MP) analyses, MrBayes v.3.0b4 [42] for Bayesian Inference (BI) analyses and MEGA v.7 [43] for Maximum Likelihood (ML) analyses. Trees resulting from the MP and BI analyses were visualized with TreeView [44]. Maximum Parsimony analyses were performed using the heuristic search option with 100 random taxon additions and subtree pruning regrafting (SPR) method as the branch-swapping algorithm. All characters were unordered and of equal weight, and gaps were treated as missing data. Maxtrees were set to 100 and branches of zero length were collapsed. Clade stability was assessed using a bootstrap analysis with 1000 replicates. The parameters including consistency index (CI), retention index (RI), tree length (TL), rescaled consistency index (RC) and homoplasy index (HI) were also calculated. Bayesian analyses employing a Markov Chain Monte Carlo sampling (MCMC) method were performed. The general time-reversible model of evolution [45], including estimation of invariable sites and assuming a gamma distribution with six rate categories was used for BI analyses. Four MCMC chains were run simultaneously, starting from random trees for 1,000,000 generations, and sampled every 100th generation for a total of 10,000 trees. The first 1000 were discarded as the burn-in phase of each analysis. Posterior probabilities (PPs) were determined from a majority-rule consensus tree generated with the remaining 9000 trees. Maximum likelihood analyses were performed on a Neighbor-Joining starting tree automatically generated by the software. Nearest-Neighbor-Interchange (NNI) was used as the heuristic method for tree inference, and 1000 bootstrap replicates were performed. MEGA v.7 was also used to determine the best nucleotide substitution model to be used for building the ML trees. The phylogenetic analyses included six well-delimitated Diaporthe species. To examine the possibility of a combined dataset, sequences from the five and four loci were aligned and combined, and the Incongruence Length Difference test was conducted in PAUP* v.4.0b10 [41]. Moreover, the highly supported clades in individual genealogies were also compared in order to detect conflict among them [33]. Individual gene trees of the D. eres species complex, comprising all available species for each locus were also constructed. These trees were rooted to Diaporthe citri and D. citrichinensis. The sequence generated in this study was deposited in GenBank (Table 1) (www.ncbi.nlm.nih.gov, accessed on 30 August 2020). The phylogenetic tree and alignments were deposited in TreeBASE (www.TreeBASE.org; Study Number S27078, accessed on 13 October 2020).
2.4. Pairwise Homoplasy Index Test and Phylogenetic Network Analysis
The concatenated five loci tree was used to infer the occurrence of sexual recombination within the D. eres complex, through the pairwise homoplasy index test (PHI) [46] implemented in SplitsTree v.4.16.1 (www.splitstree.org, accessed on 10 November 2020) [47]. To detect intragenic recombination, individual loci were also analyzed using the PHI test. Significant recombination was considered with a PHI index below 0.05 (Φw < 0.05). The relationships between closely related taxa were visualized by constructing a phylogenetic network from the concatenated dataset of five loci, using the LogDet transformation and the NeighborNet algorithm options implemented in SplitsTree v.4.16.1 [47].
2.5. Species Delimitation Analyses
To infer the species boundaries of the Diaporthe eres complex, coalescence-based methods were performed based on the combined alignment of ITS, TEF1-α, TUB2, HIS3 and CAL genes. First, the single Poisson Tree Processes model (PTP) [13], was used. The newick-format tree produced by MEGA v.7 was used for the PTP analysis with the following parameters: 500,000 MCMC generations, thinning set to 100, burn-in at 50,000 generations and conducted on the web server for PTP (http://species.h-its.org/ptp/, accessed on 24 November 2020). Second, the multi-rate PTP (mPTP), which can accommodate data sets comprised of species with different levels of molecular diversity [48], was also conducted on the web server for mPTP (http://mptp.h-its.org, accessed on 24 November 2020), using the same newick-format tree as for the single PTP analysis.
2.6. Population Genetic Diversity
To study the genetic diversity within the Diaporthe eres complex, diversity indices were calculated for each gene region and the combined datasets. The pairwise nucleotide diversity (π), haplotype diversity (hd), number of haplotypes (h), number of polymorphic sites (S) and the Tajima’s D statistical test [49] were calculated on DnaSP program v.6.12 (Barcelona, Spain) [50]. These parameters were calculated from a combined dataset of 19 ex-type species and 21 taxonomically authenticated isolates belonging to the D. eres complex (Table 1).
2.7. Morphology of the Diaporthe eres Species Complex
The alpha conidia, beta conidia and conidiophores length/width (L/W) ratios of all current species belonging to the Diaporthe eres complex were calculated. A hierarchical clustering analysis using the Ward’s method [51] was carried out, and a dendrogram was generated in R Statistical Software v.4.0.2 [52] using the DENDEXTEND package [53].
3. Results
3.1. Phylogenetic Analyses and Informative Characters
The individual gene trees of the complex (Figures S1–S5) showed that the isolates used in this study cluster in a clade containing 29 species, designated here as the D. eres species complex. The partition homogeneity test for both 5-loci and 4-loci combined alignments resulted in a low p-value (p = 0.01), indicating that the genes are unsuitable to be combined. Nevertheless, despite the observed incongruences, multilocus analyses were constructed based on five and four loci. The five loci combined alignment comprises 2364 characters (537 characters from ITS, 387 from TEF1-α, 430 from TUB2, 522 from CAL, and 488 from HIS3, including alignment gaps and indel coding); the four loci combined alignment is comprised of 1827 characters (387 from TEF1-α, 430 from TUB2, 522 from CAL, and 488 from HIS3, including alignment gaps and indel coding). Both analyses included 12 well-delimitated outgroup taxa and 40 ingroup taxa (3 from this study and 37 taxa retrieved from GenBank) (Table 1). The parsimony informative characters and the nucleotide models used for each analysis are summarized in Table 2. Moreover, sequences of the five genes were aligned and analyzed separately by Maximum Likelihood, Maximum Parsimony and Bayesian Inference analyses, and the resulting trees were compared. Only ML single trees are shown with bootstrap and posterior probabilities given for those well-supported clades (Figures S6–S10). The ML, MP and BI analyses resulted in trees that were topologically similar.
According to the informative characters provided by the Maximum Parsimony analyses, TEF1-a displayed the most informative sequences, followed by CAL, TUB2 and HIS3 loci. ITS presented the lowest percentage, indicating that this locus is unreliable for the delimitation of the D. eres species. The combined four loci dataset (TEF1-a, TUB2, CAL, and HIS3) showed better delimitation for D. eres compared to the five loci dataset (Table 2), confirming that the ITS locus is not the appropriate locus to delineate species in this genus.
3.2. Species Delimitation Based on the GCPSR Principle
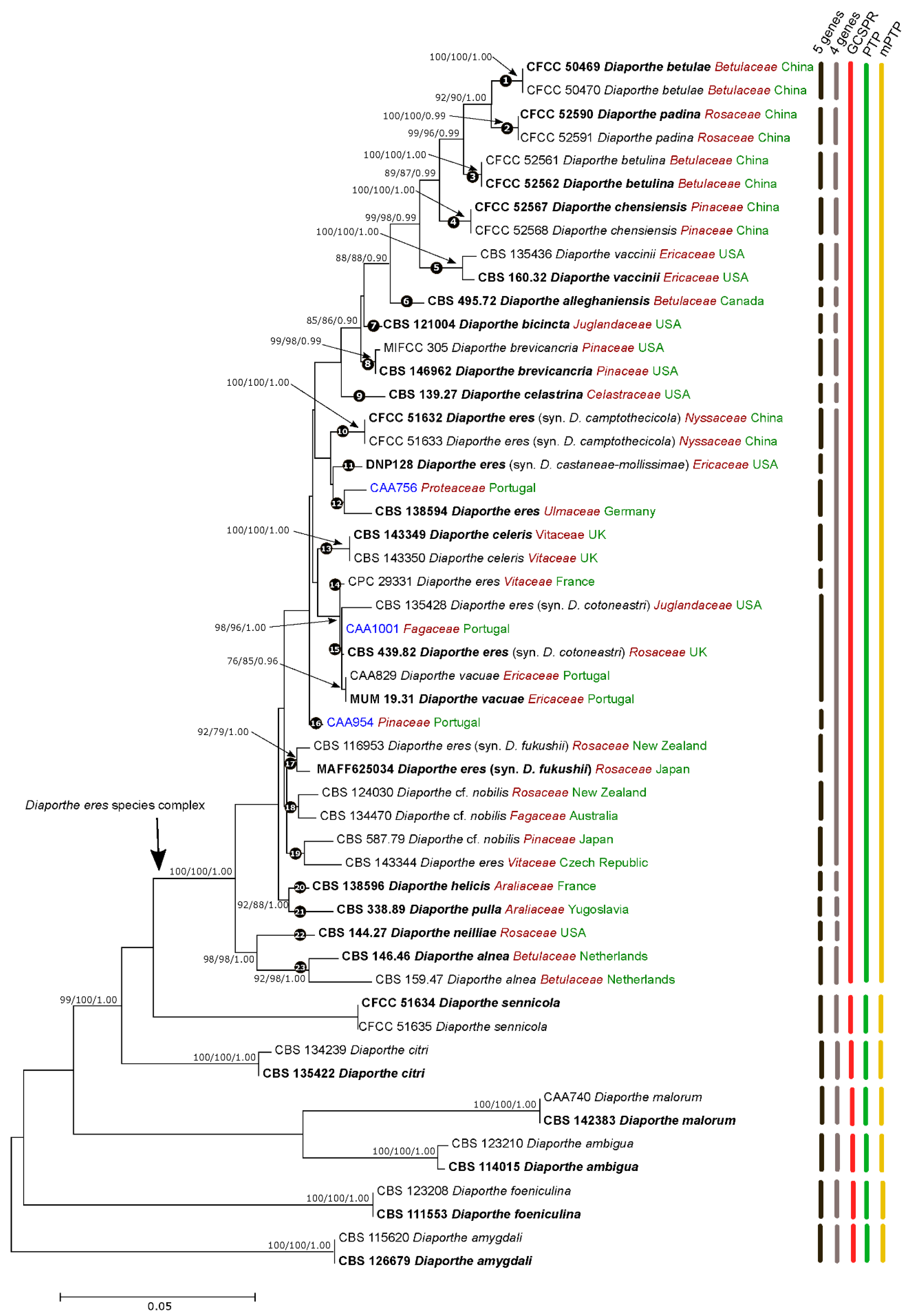
The isolates from this study clustered in a highly supported clade on both 5-loci (ML/MP/PP = 100/100/1.00) (Figure 1) and 4-loci (ML/MP/PP = 99/99/1.00) (Figure S11) phylogenies. To assess species boundaries in the D. eres complex, the GCPSR principle was applied. Our results revealed conflicts between individual phylogenies, with several nodes lacking phylogenetic support. For example, D. vaccinii is closely related to D. celeris in the TUB2 tree (Figure S8), but it is a sister species to D. camptothecicola (previously synonymized with D. eres) in the ITS individual phylogram (Figure S6). Moreover, a lack of high bootstrap and posterior probability values on both individual and combined trees in several branches were observed, revealing poor phylogenetic support among the species. It is also evident that isolates from the same species cluster in different clades in the same individual gene tree. For instance, two isolates of D. alnea are phylogenetically distant in the TUB2 tree (Figure S8), while they group together in the remaining individual phylograms. The same seems to occur with some isolates of D. eres: the isolates CBS 116953 and MAFF625034 (formerly described as D. fukushii) and two isolates of D. camptothecicola, previously synonymized with D. eres, do not cluster together in the ITS single locus tree (Figure S6). It is also notable from our analyses that the ex-type of D. alnea (CBS 146.46) is a close relative of D. allenghaniensis in the TUB2 tree (Figure S8), whereas they are different taxa in the remaining individual and combined trees. Moreover, the species D. brevicancria is phylogenetically indistinguishable from D. celastrina and D. maritima in the individual CAL tree (Figure S10). Thus, by implementing the GCPSR principle, based on the comparison of more than one gene genealogy to identify phylogenetic concordance, we verified that the node delimiting the transition from concordant branches to incongruence corresponds to the D. eres complex (Figure 1). As estimated by the initial individual trees (Figures S1–S5), the species D. maritima, D. phragmitis and D. rosicola belong to the D. eres species complex. However, given the lack of some sequences for these species, they were not included in the 4-loci and 5-loci phylogenetic analyses. However, by analyzing the individual gene trees, and given their position within the complex, we advocate that the aforementioned species should also be assigned to D. eres. Contrarily, individual gene trees are concordant regarding the six well-delimited species (D. citri, D. sennicola, D. malorum, D. foeniculina, D. ambigua and D. amygdali), indicating that there are no conflicts among the individual phylograms. This provides solid evidence that these clades represent different species.
3.3. Species Delimitation Based on Poisson Tree Processes
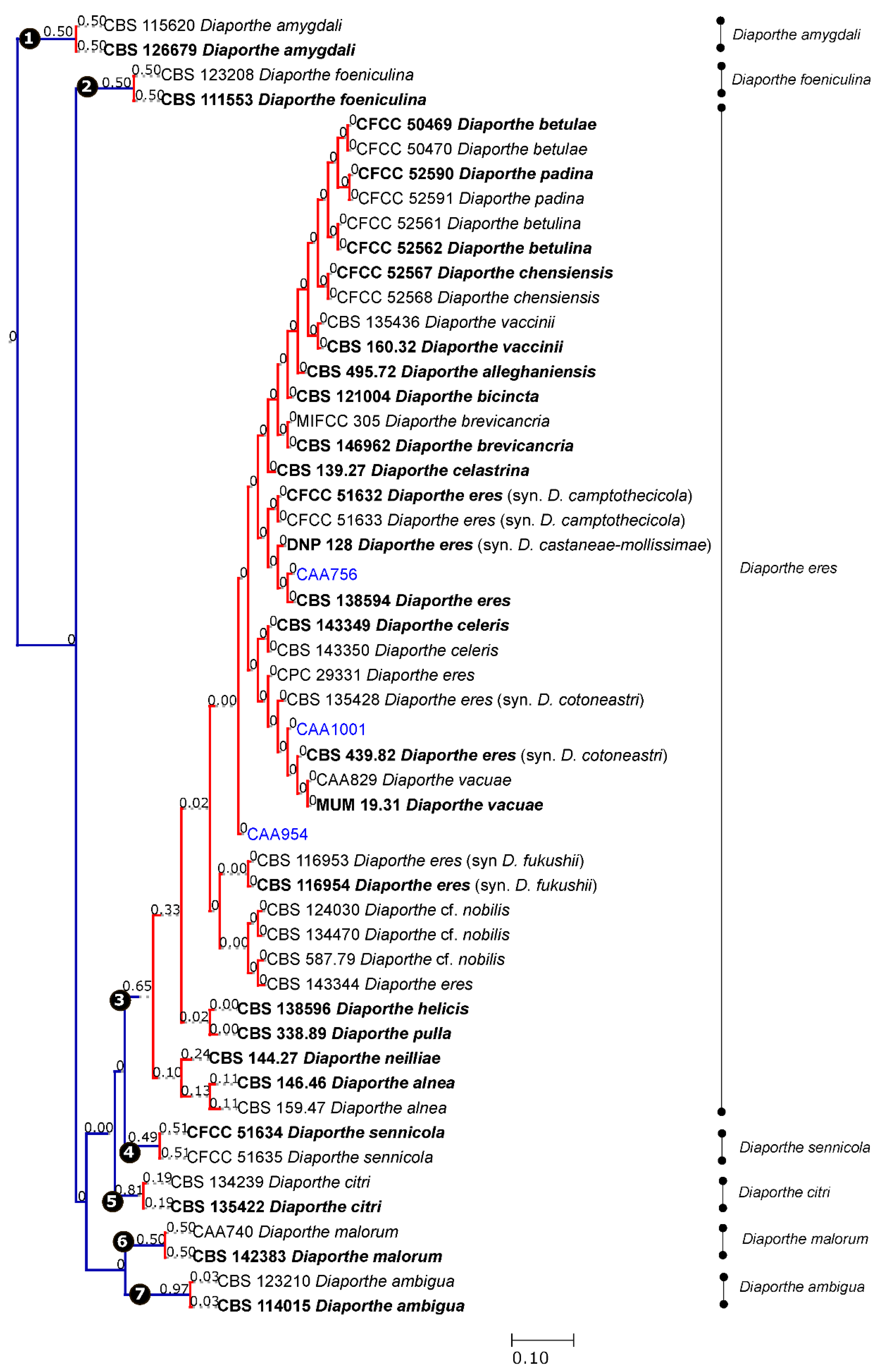
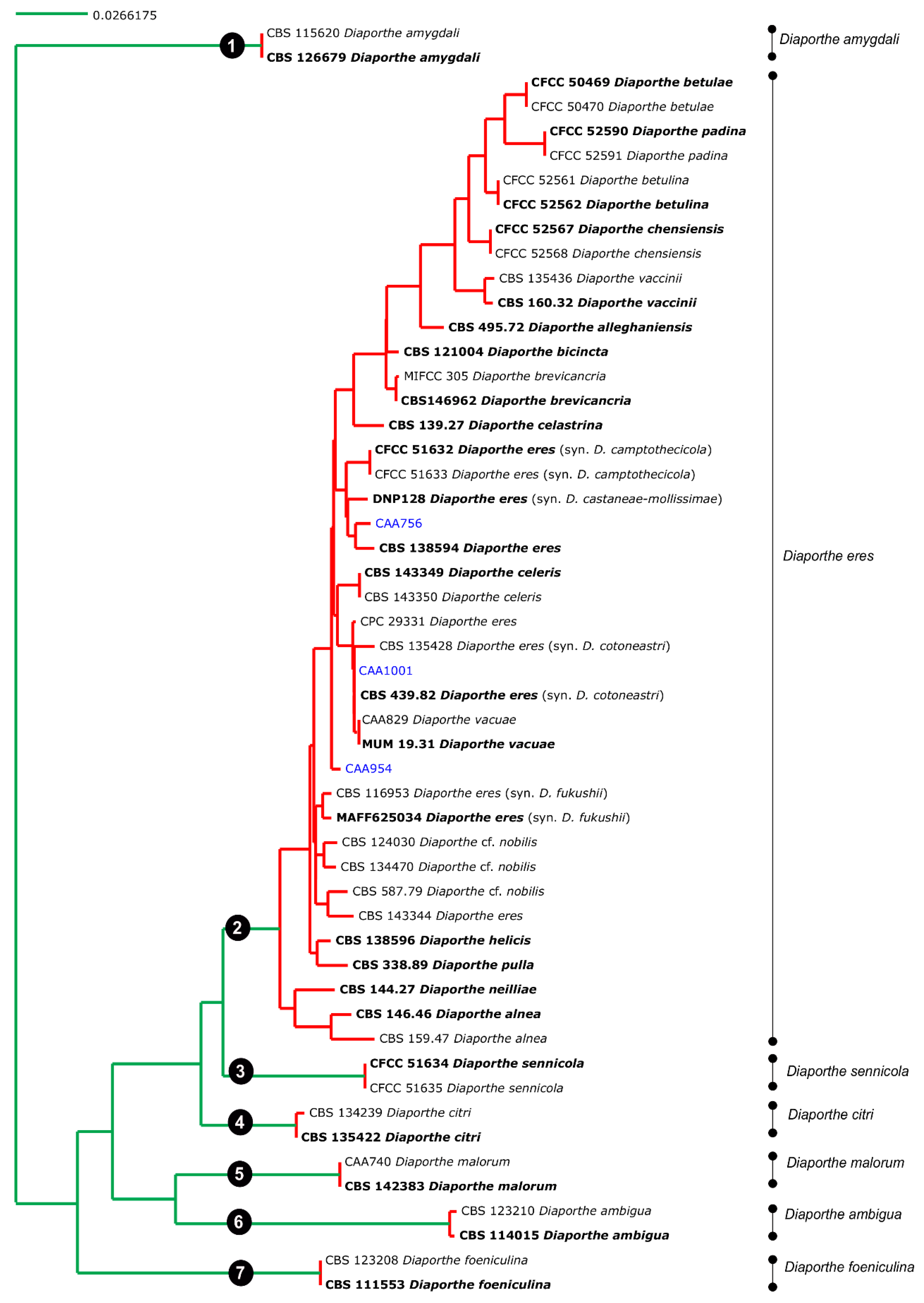
Given the lack of TEF1-α, CAL and HIS3 sequences for some species of the complex, the coalescent model applied included only those species whose five loci were available. Both single PTP and mPTP analyses gave congruent results and recognized the D. eres complex as a single species. From the analyses, the transition from blue-colored to red-colored branches (in single PTP, Figure 2), and the transition from green-colored to red-colored (in multi-rate PTP, Figure 3), evidenced that all species were comprised in one clade only, showing that the complex should be considered as a population rather than different taxa. In addition to this, a PTP analysis was performed based on the 4-loci combined alignment (Figure S12), excluding the ITS data, in order to understand whether the delimitation of species would be the same. This result corroborates the one from the PTP analysis based on the alignment of five genes, indicating that the ITS region did not affect the species delimitation, in contrast to the phylogenetic analysis. Therefore, we can consider that all species currently accepted in the D. eres complex should be recognized as a single species.
3.4. Pairwise Homoplasy Test and Phylogenetic Networks
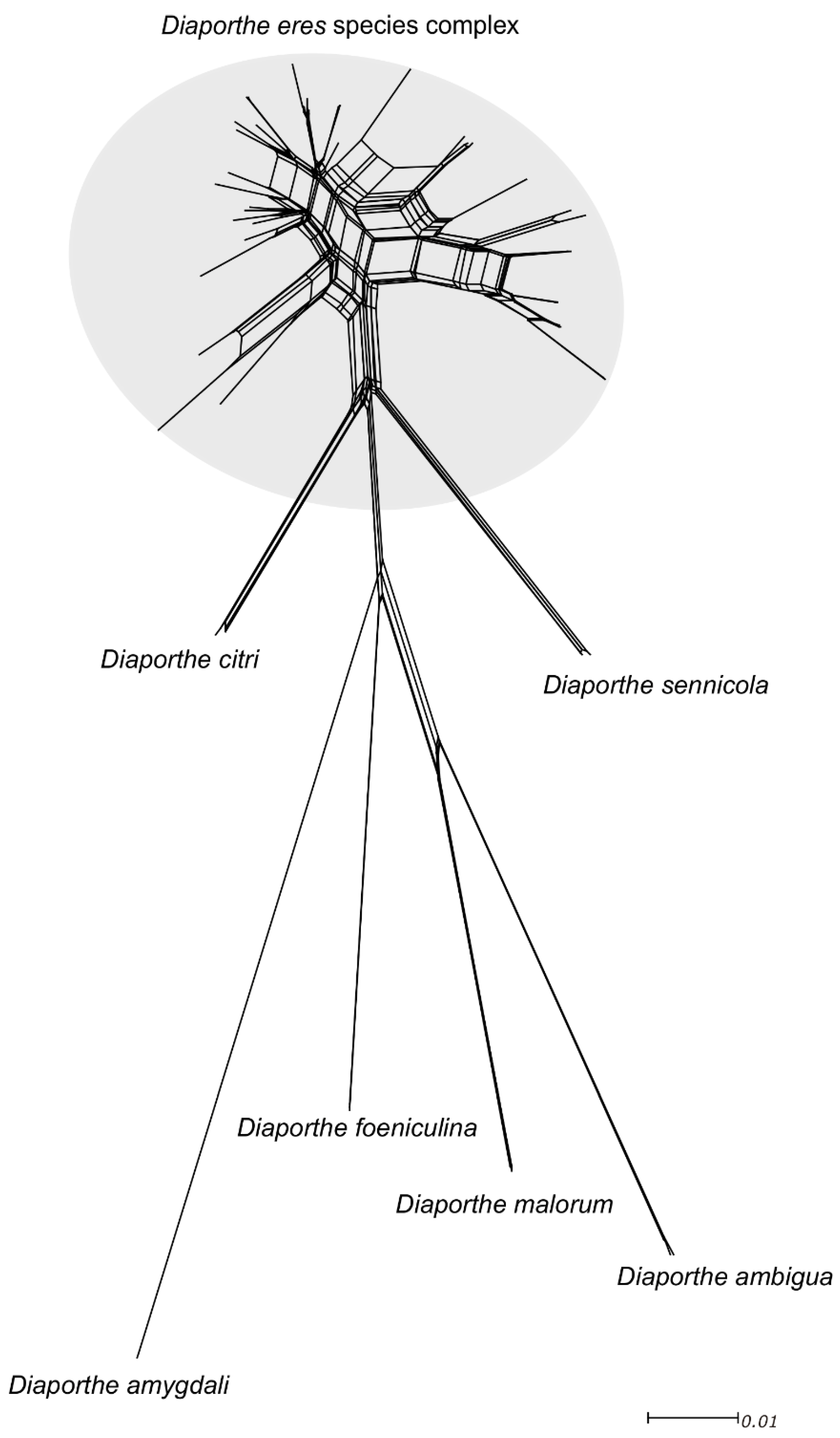
Significant recombination was detected within the D. eres complex when applying the PHI test (Φw = 0), denoting that there was no reproductive isolation within the group. Moreover, the pairwise homoplasy index test also confirmed that ITS and TUB2 loci have a significant rate of recombination (Φw = 1.098 × 10−7 and Φw = 4.175 × 10−6, respectively). Subsequently, the network relationships in the Diaporthe eres complex (Figure 4) are shown with boxes, indicating the occurrence of recombination within the group. Additionally, based on the relative distance of species and structure of the phylogenetic network, all species in the D. eres complex should be regarded as one single species.
3.5. Population Genetic Diversity
The genetic diversity data for each gene region as well as for the combined dataset is summarized in Table 3. In this analysis, all loci showed a low number of nucleotide diversity (π = 0.009 to π = 0.025) and high values for all genes (hd = 0.882 to hd = 0.966). In addition to this, TEF1-α, HIS3, TUB2, CAL and the combined five and four loci alignments did not give statistically significant negative Tajima’s D values.
3.6. Morphology of the Diaporthe eres Species Complex
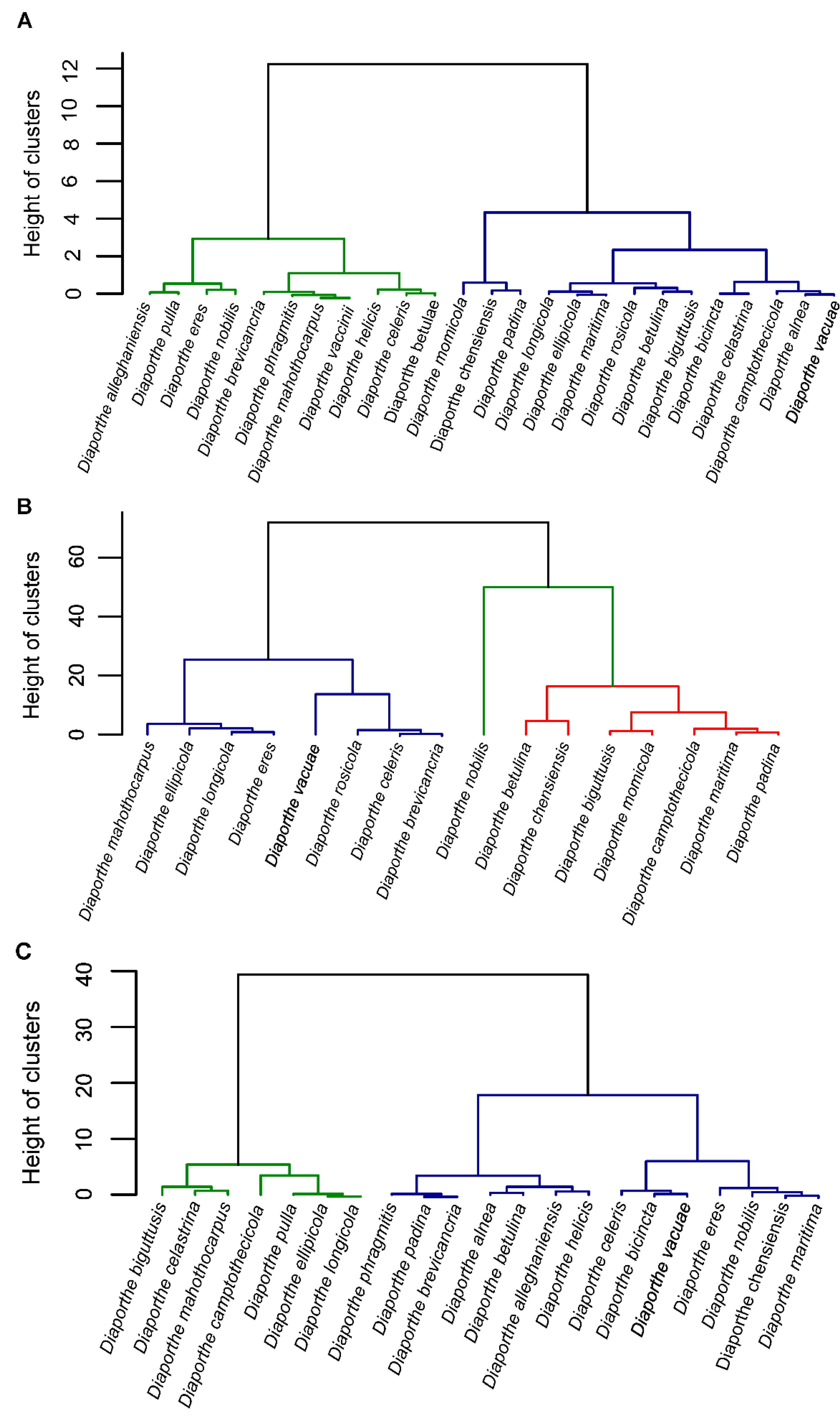
Based on published morphological descriptions of 25 species belonging to the D. eres complex (excluding D. neilliae, which lacks a taxonomic description of the asexual morph), a hierarchical clustering analysis was performed using the Ward’s method. The dendrogram was constructed based on the ratios of alpha and beta conidia as well as conidiophores length and width (Figure 5).
4. Taxonomy
The present study combined phylogenetic analyses based on the genealogical concordance principle (GCPSR), coalescence (PTP and mPTP) and morphological comparisons to delimit the species boundaries in the D. eres complex. According to the aforementioned analyses, the D. eres complex constitutes a single species rather than different lineages. In order to compare and study the micromorphological characteristics of all these species a synopsis of conidiomata, conidiogenous cells and conidia characteristics are provided in Table 4.
- Diaporthe eres Nitschke, Pyrenomycetes Germanici 2:245 (1870)
- Basionym: Phoma oblonga Desm., Annales des Sciences Naturelles; Botanique, sér. 2, 22:218 (1853)
- =Diaporthe alnea Fuckel, Jahrbücher des Nassauischen Vereins für Naturkunde 23–24:207 (1870)
- =Diaporthe pulla Nitschke, Pyrenomycetes Germanici 2:249 (1870)
- =Diaporthe helicis Niessl, Verhandlungen des naturforschenden Vereines in Brünn 16:50 (1876)
- =Diaporthe nobilis Saccardo and Spegazzini, Michelia 1:386 (1878)
- =Diaporthe bicincta (Cooke and Peck) Sacc., Sylloge fungorum (Abellini) 1:622 (1882)
- =Diaporthe neilliae Peck, Annual Report on the New York State Museum of Natural History 39:52 (1887)
- =Diaporthe celastrina (Ellis and Barthol), The Journal of Mycology 8:173 (1902)
- ≡Phomopsis oblonga (Desm.) Traverso, Fl. ital. crypt., Pars 1: Fungi. Pyrenomycetae. Xylariaceae, Valsaceae, Ceratostomataceae: 248 (1906)
- =Phomopsis velata Sacc. Traverso, Fl. ital. crypt. (Florence) 2:248 (1906)
- =Diaporthe vaccinii Shear, United States Department of Agriculture 258:7 (1931)
- =Diaporthe alleghaniensis Arnold, Canadian Journal of Botany 45:787 (1967)
- ≡Diaporthe cotoneastri Udayanga, Crous and Hyde, Fungal Diversity 56:166 (2012)
- ≡Diaporthe castaneae-mollisimae Udayanga, Crous and Hyde, Fungal Diversity 56:166 (2012)
- =Diaporthe phragmitis Crous, Fungal Planet 283:219 (2014)
- =Diaporthe biguttusis Gao and Cai, Fungal Biology 119:300 (2015)
- =Diaporthe ellipicola Gao and Cai, Fungal Biology 119:300 (2015)
- =Diaporthe longicicola Gao and Cai, Fungal Biology 119:303 (2015)
- =Diaporthe mahothocarpus Gao and Cai, Fungal Biology 119:306 (2015)
- =Diaporthe betulae Tian and Fan, Phytotaxa 269:96 (2016)
- =Diaporthe maritima Tanney, Fungal Biology 120:1454 (2016)
- =Diaporthe camptothecicola Tian and Yang, Mycotaxon 132:595 (2017)
- =Diaporthe momicola Dissanayake, Li and Hyde, Mycosphere 8:541 (2017)
- =Diaporthe fukushii Dissanayake, Phillips and Hyde, Mycosphere 8:1130 (2017)
- =Diaporthe betulina Tian and Yang, Mycokeys 39:121 (2017)
- =Diaporthe chensiensis Tian and Yang, Mycokeys 39:127 (2017)
- =Diaporthe padina Tian and Yang, Mycokeys 39:137 (2017)
- =Diaporthe celeris Guarnaccia, Woodhall and Crous, Persoonia 40:146 (2018)
- =Diaporthe rosicola Wanasinghe, Jones and Hyde, Fungal Diversity 89:187 (2018)
- =Diaporthe vacuae Hilário, Santos and Alves, Mycologia 55:207 (2020)
- =Diaporthe brevicancria Sakalidis and Medina-Mora, Phytopathology (2020)
Typification: as described by Udayanga et al. [23]—Diaporthe eres, Germany, Nordrhein-Westfalen, Munsterland, Munster Botanical Gardens, on twigs of Ulmus sp., June 1865, T. Nitschke, (lectotype B 70 0009145); Carpinion forest, on dead, attached, corticated twigs of Ulmus laevis, 5 January 2013, R. Jarling, comm. R. Schumacher (epitype BPI 892912, ex-epitype culture AR5193 = CBS 138594). Phoma oblonga, France, on twigs of Ulmus campestris, unknown collector (bound specimen of Desmazieres, Plantes Cryptogames du Nord de la France, Ed. 2, ser. 2. No. 60 in BPI). Germany, Carpinion forest, on dead, attached, corticated twigs of Ulmus laevis, 5 January 2013, R. Jarling, comm. R. Schumacher (epitype BPI 892913, ex-epitype culture AR5196 = CBS 138595). Phomopsis castaneae-mollisimae, China, Taian, Shangdong, leaf of Castanea mollissima, April 2006, S.X. Jiang (CLS 0612, holotype not seen, ex-type culture BYD1 = DNP128), ex-isotype culture BYD4 = DNP129. Diaporthe cotoneastri, UK, Scotland, Ayr, on Cotoneaster sp., May 1982, H. Butin (isotype CBS-H 7633 not seen, ex-isotype culture CBS 439.82). Phomopsis fukushii, Japan, Ibaraki, on Pyrus pyrifolia, August 1994, S. Kanematsu, (neotype BPI 892933, ex-neotype culture MAFF625034 = AR3672).
Known distribution: Austria, China, France, Germany, Italy, Japan, Korea, Latvia, Lithuania, Netherlands, New Zealand, Poland, Portugal, Russia, Serbia, South Africa, UK, USA, Yugoslavia [54].
Host range: Abutilon sp., Acer campestre, Acer nugundo, Alliaria officinalis, Allium giganteum, Arctium sp., Banksia sp., Betula alleghaniensis, Camelia sinensis, Castanea mollissima, C. sativa, Celastrus scandens, Chamaecyparis thyoides, Citrus sp., Cotoneaster sp., Cornus florida, Corylus avellena, Cucumis sp., Daphne lauriola, Eucalyptus globulus, Fraxinus excelsior, Glycine max, Hedera helix, Hordeum sp., Ilex aquifolium, Juglans cinerea, Juglans regia, Juniperus sp., Laburnum sp., Laurus nobilis, Magnolia sp., Malus pumila, M. silvestris, Opuntia sp., Osmanthus aquifolium, Oxydendrum arboreum, Phaseolus vulgaris, Picea pungens, P. glauca, P. abies, Pinus pantepylla, P. pinaster, Prunus persica, P. cerasus, P. nume, Pyrus pyrifolia, Quercus suber, Rhododendron sp., Rubus sp., Rubrus fructicosus, Rumex hydrolapathum, Salix sp., Sassafras albida, Skimmia japonica, Sorbus aucuparia, Tilis cordata, Ulmus minor, U. laevis, U. campestris, Vaccinium corymbosum, V. macrocarpon, Viburnum lantana, Vitis vinifera, Wisteria sinensis, Ziziphus jujuba [54].
Description: Both sexual and asexual morphs of D. eres have been previously described and illustrated in detail by Udayanga et al. [23].
Notes: The micromorphology of the asexual morph of all species belonging to the D. eres complex match the original description of D. eres reported by Udayanga et al. [23]: pycnidia embedded in tissue, irregularly distributed over agar surface, producing abundant and black stromata at maturity, covered in white mycelium, and oozing yellowish conidial cirrhus from ostioles. Cultural characteristics are also similar to those reported by Udayanga et al. [23]: colonies spreading on PDA in a radial pattern with white, aerial, cottony mycelium, sometimes with brown aerial mycelium at the center, becoming grey at edges of plate, and reverse white to ivory color concentric zones, becoming brownish to black with age; conidiophores, alpha conidia and beta conidia with dimensions that correlate with the ones reported by those authors. Although the asexual morph of D. neilliae was not recorded by Udayanga et al. [23], the sexual morph was compared to the one of D. eres: perithecia and ascospores of D. neilliae that match within the same ranges as those of D. eres, and asci of D. neilliae were reported to be slightly shorter than those of D. eres (45 ± 5 × 8.5 ± 0.7 μm for D. neilliae vs. 53 ± 5 × 8.0 ± 0.7 for D. eres).
5. Discussion
In recent years, the use of multilocus DNA sequence data, coupled with morphology and ecology, has been widely employed to establish robust species boundaries in the genus Diaporthe [4,66]. Nevertheless, given that several authors have identified distinct species based on the concatenation of genes, without looking for incongruences between individual gene genealogies [9], the number of species in Diaporthe has been increasing considerably. This is largely attributed to the intraspecific variability in the genus, where each clade has been incorrectly recognized as a distinct lineage [21,67].
Although the ITS region is considered the primary barcode for fungi [68], it has been argued by several authors that this ribosomal DNA region harbors a high variability, and therefore it is believed to be uninformative to resolve species within the D. eres complex [69]. Considering this, the ITS region has been excluded from the phylogenetic analysis, and currently the concatenation of TEF1-α, TUB2, HIS3 and CAL loci is widely used to resolve species in the aforementioned complex [16,18,21,23,24].
Despite Udayanga et al. [23] having proven that the concatenation of seven loci to resolve the D. eres complex, excluding the discordant ITS data, results in a robust tree congruent with the other single genes, the same was not verified in the present study. By applying the GCPSR principle, the genealogical concordance among genes must be verified [6]. However, incongruent nodes, conflicting branches, and the lack of phylogenetic support in some branches were observed in the individual phylograms versus the 4-loci and 5-loci combined trees. For example, isolates of D. eres (e.g., MAFF265034 and CFCC 51632, previously known as D. fukushii and D. camptothecicola, respectively) did not cluster together on the individual ITS tree but formed a monophyletic clade in the remaining individual and combined trees. Moreover, it was also evident that the species D. brevicancria was phylogenetically indistinguishable from D. celastrina on the individual CAL locus. Hence, by applying the genealogical concordance, we verified that the node delimiting the D. eres complex represents the transition from concordant branches to incongruence, thus indicating that this complex of species represents in fact one single species, D. eres. These incongruences observed between the individual gene genealogies confirm that the loci may harbor different evolutionary histories [70], thus making the concatenation of genes an inappropriate approach to infer the phylogenetic relationships within the D. eres complex.
Bearing in mind that the Ecological Species Concept recognizes species as of a group of individuals that occupies a specific niche [71], the use of phylogenetic approaches based on multiple loci aids in revealing ecological patterns of diversification among clades [7,72]. However, in the present study, we demonstrate that the species belonging to the D. eres complex are distributed worldwide, thus lacking a correlation between the genetic divergence of the complex and its ecological niche. Despite D. pulla being restricted to a specific locality (Yugoslavia), it was previously shown to belong to an unresolved sub-clade, which Gomes et al. [21] referred to as D. nobilis complex. Later, Udayanga et al. [23] showed that many of the isolates in the aforementioned complex clustered within D. eres, based on the GCPSR principle. Additionally, given the genetic differences between D. pulla, with its closest relative D. helicis, Udayanga et al. [23] suggested that these two species should be designated as distinct lineages. However, according to our results, D. pulla and D. helicis belong to D. eres complex and are thus synonymized here with D. eres.
Although the Morphological Species Concept was historically the dominant concept in the genus Diaporthe [19], the presence of conserved morphological characters made this concept inadequate to delineate species in this genus [20]. Based on the taxonomic description of the D. eres complex, a clear absence of morphological distinctiveness was evident. Overall, species harbor oblong to ellipsoid alpha conidia, a common presence of beta conidia, creamy to yellowish conidial cirrhus, conidiomata shapes and conidiophores dimensions that match within the same ranges. Though dendrograms of the length and width ratios distinguished the species into different groups, these are not correlated to any of the clades in the combined gene trees. Moreover, though there was higher variability in the L/W ratios observed for beta conidia, this might simply represent a character plasticity of Diaporthe rather than an indication of morphospecies. For instance, temperatures above 30 °C or the dextrose concentration (a component of the PDA medium) seems to influence the production of this type of conidia [20]. Culture characteristics are also quite identical among the recognized “species” within the D. eres complex: aerial mycelium cottony with yellowish gray to brownish-gray coloration, margin regular, producing abundant black stromata at maturity in culture and oozing yellow cirrhus. Therefore, we consider those micromorphological characters as minor differences among species, demonstrating only a high character variation within the D. eres complex. As stated by Hyde et al. [2], the recognition of significant recombination within closely related taxa should be considered as a method to justify a species. Therefore, the D. eres species complex was tested to disclose the presence of genetic recombination. Our results proved that significant genetic exchange occurs within the complex, indicating that there is no reproductive isolation between some of the putative species recognized on the 5-loci combined tree. The genetic diversity within the D. eres complex was also estimated. Low nucleotide diversity values in addition to high haplotype diversity indices indicate a high number of closely related haplotypes [73]. Moreover, although TEF1-α locus showed the highest informative characters to resolve the D. eres complex, as also corroborated by previous studies [23,25], the Tajima’s D test gave negative values for TEF1-α locus and the remaining genes (except for the ITS locus), which is indicative of inbreeding events within the population occurring at these loci. Therefore, this suggests that D. eres complex is a population that may have undergone a recent expansion, producing a large number of offspring [73,74]. In a recent study, Chaisiri et al. [25] also showed high levels of haplotype diversity within the five loci among D. eres, thus reflecting high genetic diversity. The neutrality Tajima’s test run by these authors similarly showed negative values, which suggests a population expansion in D. eres isolates. Such population expansion shown in the present study might be explained by inbreeding events among some species of the complex, occurring mainly in TUB2 and ITS loci, showing significant genetic recombination. Therefore, taking into consideration the lack of gene flow, the absence of supporting phenotypic, geographic, and ecological differences, the recent divergence and the possibility of incomplete lineage sorting of the D. eres complex may be considered as ongoing evolving lineages [75].
Given the existence of conflicts between individual gene trees, the impossibility to combine genes and the lack of phenotypic distinctiveness, we attempted to delimit the species boundaries of the D. eres species complex through coalescent-based models. This latter approach involves understanding how several species are related by modeling the genealogical history of individuals with a common ancestor [7,13,76]. The General Mixed-Yule Coalescent (GMYC) and the Poisson Tree Processes (PTPs) models are widely used to identify branching patterns between divergence and intraspecific diversification and thus to distinguish between species and populations [77]. However, the GMYC model can overestimate the number of taxa, particularly in species with a strong intraspecific genetic structure [75]. An important advantage of using PTP analyses (both single and multi-rate) is that it models speciation events in terms of the number of nucleotide substitutions [13]. Therefore, it avoids the computation-intensive process of generating time-calibrated ultrametric trees, which are required as an input for GMYC analysis. Another drawback of this latter analysis is the choice of the molecular clock, which is essential to infer the timing of evolutionary divergence events in a given phylogeny [78]. However, this is somehow difficult to predict, as fossil evidence remains scarce for fungi [79]. For this reason, PTP analysis is considered to yield more accurate delimitations than GMYC [80], so this was the one adopted in the present study. According to our results, the highly supported clades recognized as distinct species in the combined 5-loci and 4-loci trees were not concordant with the coalescent methods, as both single and multi-rate PTP analyses inferred that D. eres complex should be recognized as a single species. Furthermore, based on the phylogenetic network performed, it is notable that the species within the D. eres complex are linked with boxes, which is indicative of the presence of recombination. For this reason, we considered that all species falling into the D. eres complex should be regarded as one single species, i.e., D. eres, rather than different taxa. According to Yang et al. [18], the species D. maritima, D. phragmitis and D. rosicola belong to the D. eres species complex. Given the lack of sequences available for some loci, these species could not be included in the analyses. Nevertheless, considering that these species fall into the D. eres complex, based in the individual genealogies performed, and knowing that Chasiri et al. [25] synonymized D. rosicola as D. eres, which we also corroborate in the present study, we feel comfortable considering D. maritima and D. phragmitis as synonymous with D. eres.
Of particular relevance is the synonymization of D. vaccinii with D. eres. Diaporthe vaccinii has been regarded as a common and important pathogen of the Vaccinium species, especially in the US, where it was originally reported [26]. In Europe, it is a quarantine organism and is regarded as eradicated from all countries where it was previously detected [81]. We recognize the potential impact that our proposal may have in the plant pathology community, but the results from the integrative approach performed in this study provide strong evidence that D. vaccinii cannot be regarded as a distinct species from D. eres. Previous studies recognize that both species are morphologically indistinguishable and phylogenetically very closely related [16,17,23,24,26]. Thus, it is not unreasonable to accept that they represent a single species.
The species D. vaccinii was described in an epoch where host association was regarded as an important character to delimit species. It is now widely accepted that most (if not all) species of Diaporthe are not host specific [19,20], and D. vaccinii is one of the rare exceptions still accepted. Recognizing it as a synonym of D. eres means that its status as a major pathogen of blueberry would need to be reassessed. Previous studies [82] have suggested that D. vaccinii is probably not a major threat to blueberry production in Europe and that its status as a quarantine organism should be reappraised.
Following the most common species concept used in fungi, the Phylogenetic Species Concept (PSC) [6], a species is assigned to a phylogenetic cluster that shares a most recent common ancestor, and it differs phenotypically from its closest relatives [8,10]. However, this is not always observed in the genus Diaporthe, as in the case of the Diaporthe eres complex, which was shown in this study to display little or no morphological variation. Moreover, the incongruences observed among the individual genealogies make the concatenation of multiple loci an inappropriate approach to delimit species. For this reason, we believe that the genealogical concordance allied to the recognition of significant recombination among species must be applied in future studies to delimit the species boundaries in the genus Diaporthe.
6. Conclusions
In the present study, phylogenetic analyses based on the GCPSR principle and the coalescent-based species model, PTP, prove that the D. eres complex is a population with evolving lineages, rather than a complex composed of distinct species. Furthermore, the pairwise homoplasy index test and the comparison of morphological and ecological characters highlight the absence of gene flow within the population, given that there is no evidence of reproductive isolation or of geographical barriers. This study suggests that the identification of species in Diaporthe has been largely overestimated, since the use of multilocus DNA sequences has been widely used without comparing the individual genealogies to look for incongruent nodes. This is particularly important in the genus Diaporthe, given the presence of a high intraspecific variability that might have been erroneously regarded as an aspect to describe novel taxa. Hence, individual gene genealogies must always be compared to look for incongruences among them. Once incongruent branches or a lack of phylogenetic support is observed, careful assumptions need to be made prior the description of new species in the genus Diaporthe. We also recommend that several strains from different locations should be included in the analyses, whenever possible, in the attempt to assess the intraspecific variation. Moreover, bearing in mind that the ITS region is the primary barcode for fungi, and it has been adopted as the genetic marker of choice for species delimitation, we advocate that this ribosomal region should not be excluded from the phylogenetic analyses, but carefully analyzed along with the other protein coding genes used in the genus Diaporthe, such as TEF1-α, TUB2, HIS3 and CAL loci. In addition, further studies based on the coalescent models should also be implemented in the genus Diaporthe to provide stronger support to infer the phylogenetic relationships between cryptic species. Large-scale whole genome sequencing must also be considered in the future to provide insights into the validity of the current five loci used for molecular identification in the genus Diaporthe, as well as to identify new markers to be used in the delimitation of species in this genus or even to develop a genome-based taxonomy approach to delimit species in the genus.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jof7070507/s1, Figures S1–S5. Maximum Likelihood trees based on ITS, TEF1-α, TUB2, HIS3 and CAL sequence data, respectively, for all species of the Diaporthe eres species complex. Figures S6–S10. Maximum Likelihood tree based on ITS, TEF1-α, TUB2, HIS3 and CAL sequence data, respectively, for the Diaporthe eres species complex and related species. Figure S11. Maximum Likelihood tree based on TEF1-α, TUB2, HIS3 and CAL sequence data for the Diaporthe eres species complex and related species. Figure S12. Results of the single PTP analyses for the Diaporthe eres species complex and related taxa, based on TEF1-α, TUB2, HIS3 and CAL loci on Bayesian and Maximum Likelihood topologies.
Author Contributions
Conceptualization, S.H. and A.A.; methodology, S.H. and A.A.; formal analysis, S.H. and M.F.M.G.; investigation, S.H.; resources, A.A.; data curation, S.H. and M.F.M.G.; writing—original draft preparation, S.H.; writing—review and editing, S.H., M.F.M.G. and A.A.; supervision, A.A.; project administration, A.A.; funding acquisition, A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Portuguese Foundation for Science and Technology (FCT/MCTES) that finances CESAM (Centre for Environmental and Marine Studies) (UIDP/50017/2020 + UIDB/50017/2020) through national funds, and the PhD grants of Sandra Hilário (SFRH/BD/137394/2018) and Micael Gonçalves (SFRH/BD/129020/2017).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed in this study are included in this article and its supplementary information files. All sequence data are available in the NCBI GenBank, following the accession numbers in the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wingfield, M.J.; De Beer, Z.W.; Slippers, B.; Wingfield, B.D.; Groenewald, J.Z.; Lombard, L.; Crous, P.W. One fungus, one name promotes progressive plant pathology. Mol. Plant Pathol. 2012, 13, 604–613. [Google Scholar] [CrossRef]
- Hyde, K.D.; Jeewon, R.; Chen, Y.J.; Bhunjun, C.S.; Calabon, M.S.; Jiang, H.B.; Lin, C.-G.; Norphanphoun, C.; Sysouphathong, P.; Pem, D.; et al. The numbers of fungi: Is the descriptive curve flattening? Fungal Divers. 2020, 103, 219–271. [Google Scholar] [CrossRef]
- Lücking, R.; Hawksworth, D.L. Formal description of sequence-based voucherless Fungi: Promises and pitfalls, and how to resolve them. IMA Fungus 2018, 9, 143–165. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.H.; Liu, F.; Duan, W.; Crous, P.W.; Cai, L. Diaporthe is paraphyletic. IMA Fungus 2017, 8, 163–187. [Google Scholar] [CrossRef]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinia, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Wang, M.; Damm, U.; Crous, P.W.; Cai, L. Species boundaries in plant pathogenic fungi: A Colletotrichum case study. BMC Evol. Biol. 2016, 16, 81. [Google Scholar] [CrossRef] [Green Version]
- Inderbitzin, P.; Robbertse, B.; Schoch, C.L. Species identification in plant-associated prokaryotes and fungi using DNA. Phytobiomes J. 2020, 4, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.E.; Timmer, L.W.; Lawrence, C.B.; Pryor, B.M.; Peever, T.L. Discord between morphological and phylogenetic species boundaries: Incomplete lineage sorting and recombination results in fuzzy species boundaries in an asexual fungal pathogen. BMC Evol. Biol. 2014, 14, 38. [Google Scholar] [CrossRef]
- Achari, S.R.; Kaur, J.; Dinh, Q.; Mann, R.; Sawbridge, T.; Summerell, B.A.; Edwards, J. Phylogenetic relationship between Australian Fusarium oxysporum isolates and resolving the species complex using the multispecies coalescent model. BMC Genom. 2020, 21, 248. [Google Scholar] [CrossRef] [Green Version]
- Rannala, B.; Yang, Z. Species Delimitation. In Phylogenetics in the Genomic Era; Scornavacca, C., Delsuc, F., Galtier, N., Eds.; No commercial publisher; 2020; Chapter 5.5; pp. 1–18. Available online: https://hal.inria.fr/PGE (accessed on 12 September 2020).
- Carstens, B.C.; Knowles, L.L. Estimating species phylogeny from gene-tree probabilities despite incomplete lineage sorting: An example from Melanoplus grasshoppers. Syst. Biol. 2007, 56, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, D.E.; Oliva, M.; Leiva, S.; Mendoza, J.E.; Bobadilla, L.; Angulo, G.; Calderon, M.S. Phylogeny and species delimitations in the entomopathogenic genus Beauveria (Hypocreales, Ascomycota), including the description of B. peruviensis sp. nov. MycoKeys 2019, 58, 47–68. [Google Scholar] [CrossRef]
- Hilário, S.; Santos, L.; Alves, A. Diaporthe amygdali, a species complex or a complex species? Fungal Biol. 2021, 125, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Yang, Q.; Bezerra, J.D.; Alvarez, L.V.; Tian, C. Diaporthe from walnut tree (Juglans regia) in China, with insight of the Diaporthe eres complex. Mycol. Prog. 2018, 17, 841–853. [Google Scholar] [CrossRef]
- Guarnaccia, V.; Groenewald, J.Z.; Woodhall, J.; Armengol, J.; Cinelli, T.; Eichmeier, A.; Ezra, D.; Fontaine, F.; Gramaje, D.; Gutierrez-Aguirregabiria, A.; et al. Diaporthe diversity and pathogenicity revealed from a broad survey of grapevine diseases in Europe. Persoonia 2018, 40, 135–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Fan, X.L.; Guarnaccia, V.; Tian, C.M. High diversity of Diaporthe species associated with dieback diseases in China, with twelve new species described. MycoKeys 2018, 39, 97–149. [Google Scholar] [CrossRef] [Green Version]
- Rehner, S.A.; Uecker, F.A. Nuclear ribosomal internal transcribed spacer phylogeny and host diversity in the coelomycete Phomopsis. Can. J. Bot. 1994, 72, 1666–1674. [Google Scholar] [CrossRef]
- Mostert, L.; Crous, P.W.; Kang, J.C.; Phillips, A.J.L. Species of Phomopsis and a Libertella sp. occurring on grapevines with specific reference to South Africa: Morphological, cultural, molecular and pathological characterization. Mycologia 2001, 93, 146–167. [Google Scholar]
- Gomes, R.R.; Glienke, C.; Videira, S.I.R.; Lombard, L.; Groenewald, J.Z.; Crous, P.W. Diaporthe: A genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 2013, 31, 1–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Felix, Y.; Hernández-Restrepo, M.; Wingfield, M.J.; Akulov, A.; Carnegie, A.J.; Cheewangkoon, R.; Gramaje, D.; Groenewald, J.Z.; Guarnaccia, V.; Halleen, F.; et al. Genera of phytopathogenic fungi: GOPHY 2. Stud. Mycol. 2019, 92, 47–133. [Google Scholar] [CrossRef] [PubMed]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Chukeatirote, E.; Hyde, K.D. Insights into the genus Diaporthe: Phylogenetic species delimitation in the D. eres species complex. Fungal Divers. 2014, 67, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.S.; Crous, P.W.; Bai, Q.; Fu, M.; Yang, M.M.; Wang, X.H.; Du, Y.M.; Hong, N.; Xu, W.X.; Wang, G.P. High diversity of Diaporthe species associated with pear shoot canker in China. Persoonia 2020, 45, 132–162. [Google Scholar] [CrossRef]
- Chaisiri, C.; Liu, X.; Lin, Y.; Fu, Y.; Zhu, F.; Luo, C. Phylogenetic and haplotype network analyses of Diaporthe eres species in China based on sequences of multiple loci. Biology 2021, 10, 179. [Google Scholar] [CrossRef]
- Lombard, L.; van Leeuwen, G.C.M.; Guarnaccia, V.; Polizii, G.; van Rijswick, P.C.J.; Rosendahl, C.H.M.; Gabler, J.; Crous, P.W. Diaporthe species associated with Vaccinium, with specific reference to Europe. Phytopathol. Mediterr. 2014, 53, 287–299. [Google Scholar]
- Ali, S.; Renderos, W.; Bevis, E.; Hebb, J.; Abbasi, P.A. Diaporthe eres causes stem cankers and death of young apple rootstocks in Canada. Can. J. Plant Pathol. 2020, 42, 218–227. [Google Scholar] [CrossRef]
- Lopes, A.F.; Batista, E.; Hilário, S.; Santos, L.; Alves, A. Occurrence of Diaporthe species in Eucalyptus globulus, Pinus pinaster and Quercus suber in Portugal. For. Pathol. 2021, 51, e12674. [Google Scholar]
- Hilário, S.; Amaral, I.; Gonçalves, F.M.; Lopes, A.; Santos, L.; Alves, A. Diversity and pathogenicity of Diaporthe species on blueberry plants in Portugal, with description of 4 new species. Mycologia 2020, 112, 293–308. [Google Scholar] [CrossRef]
- Möller, E.M.; Bahnweg, G.; Sandermann, H.; Geiger, H.H. A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res. 1992, 20, 6115–6116. [Google Scholar] [CrossRef] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Alves, A.; Correia, A.; Luque, J.; Phillips, A.J.L. Botryosphaeria corticola, sp. nov. on Quercus species, with notes and description of Botryosphaeria stevensii and its anamorph, Diplodia mutila. Mycologia 2004, 96, 598–613. [Google Scholar] [CrossRef]
- Alves, A.; Crous, P.W.; Correia, A.; Phillips, A.J.L. Morphological and molecular data reveal cryptic speciation in Lasiodiplodia theobromae. Fungal Divers. 2008, 28, 1–13. [Google Scholar]
- Glass, N.L.; Donaldson, G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous Ascomycetes. Appl. Environ. Microbiol. 1995, 61, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, K.; Cigelnik, E. Two divergent intragenomic rDNA ITS2 types within a monophyletic lineage of the fungus Fusarium are nonorthologous. Mol. Phylogenet. Evol. 1997, 7, 103–116. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z.; Risede, J.M.; Hywe, N.L. Calonectria species and their Cylindrocladium anamorphs: Species with sphaeropedunculate vesicles. Stud. Mycol. 2004, 50, 415–430. [Google Scholar]
- Carbone, I.; Kohn, L.M. A method for designing primer set for speciation studies in filamentous ascomycetes. Mycologia 1999, 91, 553–556. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Higgins, D.G. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Vaydia, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the last assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP* Phylogenetic Analysis Using Parsimony, (*and Other Methods); Version 4.0 b10; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Page, R.D.M. TreeView: An application to display phylogenetic trees on personal computers. CABIOS 1996, 12, 357–358. [Google Scholar]
- Rodríguez, F.; Oliver, J.L.; Marín, A.; Medina, J.R. The general stochastic model of nucleotide substitutions. J. Theor. Biol. 1990, 142, 485–501. [Google Scholar] [CrossRef]
- Bruen, T.C. A simple and robust statistical test for detecting the presence of recombination. Genetics 2005, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Kapli, P.; Lutteropp, S.; Zhang, J.; Kobert, K.; Pavlidis, P.; Stamatakis, A.; Flouri, T. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 2017, 33, 1630–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Murtagh, F.; Legendre, P. Ward’s hierarchical agglomerative clustering method: Which algorithms implement Ward’s criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. Available online: https://www.R-project.org/ (accessed on 14 January 2021).
- Galili, T. Dendextend: An R package for visualizing, adjusting, and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [Green Version]
- Farr, D.F.; Rossman, A.Y. Fungal Databases, U.S. National Fungus Collections, ARS, USDA. 2021. Available online: https://nt.ars-grin.gov/fungaldatabases/ (accessed on 8 April 2021).
- Du, Z.; Fan, X.L.; Hyde, K.D.; Yang, Q.; Liang, Y.M.; Tian, C.M. Phylogeny and morphology reveal two new species of Diaporthe from Betula spp. in China. Phytotaxa 2016, 269, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.H.; Su, Y.Y.; Sun, W.; Cai, L. Diaporthe species occurring on Lithocarpus glabra in China, with descriptions of five new species. Fungal Biol. 2015, 119, 295–309. [Google Scholar] [CrossRef]
- Sakalidis, M.; Medina-Mora, C.M.; Shin, K.; Fulbright, D.W. Characterization of Diaporthe spp. associated with spruce decline on Colorado blue spruce in Michigan. Phytopathology 2021, 111, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Fan, X.L.; Du, Z.; Liang, Y.M.; Tian, C.M. Diaporthe camptothecicola sp. nov. on Camptotheca acuminata in China. Mycotaxon 2017, 132, 591–601. [Google Scholar] [CrossRef]
- Gao, Y.H.; Sun, W.; Su, Y.Y.; Cai, L. Three new species of Phomopsis in Gutianshan nature reserve in China. Mycol. Prog. 2014, 13, 111–121. [Google Scholar] [CrossRef]
- Tanney, J.B.; McMullin, D.R.; Green, B.D.; Miller, J.D.; Seifert, K.A. Production of antifungal and antiinsectan metabolites by the Picea endophyte Diaporthe maritima sp. nov. Fungal Biol. 2016, 120, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, A.J.; Zhang, W.; Liu, M.; Hyde, K.D.; Zhao, W.S.; Li, X.H.; Yan, J.Y. Diaporthe species associated with peach tree dieback in Hubei, China. Mycosphere 2017, 8, 533–549. [Google Scholar]
- Li, Y.; Tan, P.; Zhao, D.G. Diaporthe nobilis, a new record on Camellia sinensis in Guizhou Province, China. Mycosphere 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Crous, P.W.; Wingfield, M.J.; Schumacher, R.K.; Summerell, B.A.; Giraldo, A.; Gené, J.; Guarro, J.; Wanasinghe, D.N.; Hyde, K.D.; Camporesi, E.; et al. Fungal Planet description sheets: 281–319. Persoonia 2014, 33, 212–289. [Google Scholar] [CrossRef]
- Wanasinghe, D.N.; Phukhamsakda, C.; Hyde, K.D.; Jeewon, R.; Lee, H.B.; Jones, E.G.; Tibpromma, S.; Tennakoon, D.S.; Jayasiri, S.C.; Gafforov, Y.; et al. Fungal diversity notes 709–839: Taxonomic and phylogenetic contributions to fungal taxa with an emphasis on fungi on Rosaceae. Fungal Divers. 2018, 89, 1–236. [Google Scholar] [CrossRef]
- Farr, D.F.; Castlebury, L.A.; Rossman, A.Y. Morphological and molecular characterization of Phomopsis vaccinii and additional isolates of Phomopsis from blueberry and cranberry in the eastern United States. Mycologia 2002, 94, 494–504. [Google Scholar] [CrossRef]
- Santos, L.; Alves, A.; Alves, R. Evaluating multi-locus phylogenies for species boundaries determination in the genus Diaporthe. Peer J. 2017, 5, e3120. [Google Scholar] [CrossRef] [Green Version]
- Udayanga, D.; Liu, X.; McKenzie, E.H.C.; Chukeatirote, E.; Bahkali, A.H.A.; Hyde, K.D. The genus Phomopsis: Biology, applications, species concepts and names of common phytopathogens. Fungal Divers. 2011, 50, 189–225. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Crouch, J.A.; Clarke, B.B.; Hillman, B.I. What is the value of ITS sequence data in Colletotrichum systematics and species diagnosis? A case study using the falcate-spored graminicolous Colletotrichum group. Mycologia 2009, 101, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenkamp, E.T.; Wingfield, M.J.; McTaggart, A.R.; Wingfield, B.D. Fungal species and their boundaries matter—Definitions, mechanisms and practical implications. Fungal Biol. Rev. 2018, 32, 104–116. [Google Scholar] [CrossRef]
- Gazis, R.; Rehner, S.; Chaverri, P. Species delimitation in fungal endophyte diversity studies and its implications in ecological and biogeographic inferences. Mol. Ecol. 2011, 20, 3001–3013. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Harclerode, F.M.; Strauss, R.E.; Fulhorst, C.F.; Milazzo, M.L.; Ruthven, D.C.; Bradley, R.D. Molecular evidence for high levels of intrapopulation genetic diversity in woodrats (Neotoma micropus). J. Mammal. 2007, 88, 360–370. [Google Scholar] [CrossRef] [Green Version]
- Manawasinghe, I.S.; Dissanayake, A.; Liu, M.; Wanasinghe, D.; Xu, J.; Zhao, W.; Zhang, W.; Zhou, Y.; Hyde, K.D.; Brooks, S.; et al. High genetic diversity and species complexity of Diaporthe associated with grapevine dieback in China. Front. Microbiol. 2019, 10, 1936. [Google Scholar] [CrossRef]
- Boluda, C.G.; Rico, V.J.; Divakar, P.K.; Nadyeina, O.; Myllys, L.; McMullin, R.T.; Zamora, J.C.; Scheidegger, C.; Hawksworth, D.L. Evaluating methodologies for species delimitation: The mismatch between phenotypes and genotypes in lichenized fungi (Bryoria sect. Implexae, Parmeliaceae). Persoonia 2019, 42, 75–100. [Google Scholar] [CrossRef] [Green Version]
- Pons, J.; Barraclough, T.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.; Hazell, S.; Kamoun, S.; Sumlin, W.; Vogler, A. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Matute, D.R.; Sepúlveda, V.E. Fungal species boundaries in the genomics era. Fungal Genet. Biol. 2019, 131, 103249. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.; Duchêne, S. Molecular-clock methods for estimating evolutionary rates and timescales. Mol. Ecol. 2014, 23, 5947–5965. [Google Scholar] [CrossRef] [PubMed]
- Berbee, M.L.; Taylor, J.W. Dating the molecular clock in fungi—How close are we? Fungal Biol. Rev. 2010, 24, 1–16. [Google Scholar] [CrossRef]
- Tang, C.Q.; Humphreys, A.M.; Fontaneto, D.; Barraclough, T.G.; Paradis, E. Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol. Evol. 2014, 5, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Jeger, M.; Bragard, C.; Caffier, D.; Candresse, T.; Chatzivassiliou, E.; Dehnen-Schmutz, K.; Gilioli, G.; Grégoire, J.C.; Miret, J.; MacLeod, A.; et al. Pest risk assessment of Diaporthe vaccinii for the EU territory. EFSA J. 2017, 15, 1–185. [Google Scholar]
- Cardinaals, J.; Wenneker, M.; Voogd, J.G.B.; van Leeuwen, G.C.M. Pathogenicity of Diaporthe spp. on two blueberry cultivars (Vaccinium corymbosum). EPPO Bull. 2018, 48, 128–134. [Google Scholar] [CrossRef]
Figure 1.
Maximum Likelihood (ML) tree of the Diaporthe eres complex and related species, based on ITS TEF1-α, TUB2, HIS3 and CAL loci. The ML tree is drawn to scale and was constructed based on the Tamura-Nei parameter model, assuming a gamma distribution. ML and MP bootstrap values greater than 70% and posterior probabilities (PPs) greater than 0.80 are shown at the nodes. The ex-type strains are in bold. The isolates from this study are indicated in blue. Species name is followed by the family of the host it was isolated from (red) and country of origin (green).
Figure 1.
Maximum Likelihood (ML) tree of the Diaporthe eres complex and related species, based on ITS TEF1-α, TUB2, HIS3 and CAL loci. The ML tree is drawn to scale and was constructed based on the Tamura-Nei parameter model, assuming a gamma distribution. ML and MP bootstrap values greater than 70% and posterior probabilities (PPs) greater than 0.80 are shown at the nodes. The ex-type strains are in bold. The isolates from this study are indicated in blue. Species name is followed by the family of the host it was isolated from (red) and country of origin (green).

Figure 2.
Results of the single PTP analyses for the Diaporthe eres species complex and related taxa, based on Bayesian and Maximum Likelihood topologies. Putative species clusters are indicated using transitions from blue-colored to red-colored branches and represented by circles (1–7). The isolates obtained in this study are indicated in blue.
Figure 2.
Results of the single PTP analyses for the Diaporthe eres species complex and related taxa, based on Bayesian and Maximum Likelihood topologies. Putative species clusters are indicated using transitions from blue-colored to red-colored branches and represented by circles (1–7). The isolates obtained in this study are indicated in blue.

Figure 3.
Results of the mPTP analysis for the Diaporthe eres species complex and related taxa. Putative species clusters are indicated using transitions from green-colored to red-colored branches. The isolates obtained in this study are indicated in blue.
Figure 3.
Results of the mPTP analysis for the Diaporthe eres species complex and related taxa. Putative species clusters are indicated using transitions from green-colored to red-colored branches. The isolates obtained in this study are indicated in blue.

Figure 4.
Phylogenetic network from the concatenated data (ITS, TEF1-α, TUB2, HIS3 and CAL) representing the structure of the Diaporthe eres species complex and other well-delimitated species, based on the LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position.
Figure 4.
Phylogenetic network from the concatenated data (ITS, TEF1-α, TUB2, HIS3 and CAL) representing the structure of the Diaporthe eres species complex and other well-delimitated species, based on the LogDet transformation and the NeighborNet algorithm, inferred by SplitsTree. The scale bar represents the expected number of substitutions per nucleotide position.

Figure 5.
Dendrogram from the hierarchical clustering analysis based on the Ward’s method showing the distribution of the length and width ratio (L/W) of alpha conidia (A), beta conidia (B) and conidiophores (C) of all species isolates from the Diaporthe eres complex. Green, blue and red colors represent the different clusters based on the micromorphological descriptions.
Figure 5.
Dendrogram from the hierarchical clustering analysis based on the Ward’s method showing the distribution of the length and width ratio (L/W) of alpha conidia (A), beta conidia (B) and conidiophores (C) of all species isolates from the Diaporthe eres complex. Green, blue and red colors represent the different clusters based on the micromorphological descriptions.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
List of Diaporthe species used in this study.
| Species | Strain 1 | Host | Country | GenBank Accession | ||||
|---|---|---|---|---|---|---|---|---|
| ITS | TEF1-α | TUB2 | HIS3 | CAL | ||||
| D. ambigua | CBS 114015 | Pyrus communis | South Africa | KC343010 | KC343736 | KC343978 | KC343494 | KC343252 |
| CBS 123210 | Foeniculum vulgare | Portugal | KC343012 | KC343738 | KC343980 | KC343496 | KC343254 | |
| D. amygdali | CBS 126679 | Prunus dulcis | Portugal | KC343022 | KC343748 | KC343990 | KC343506 | KC343264 |
| CBS 115620 | Prunus persica | USA | KC343020 | KC343746 | KC343988 | KC343504 | KC343262 | |
| D. citri | CBS 134239 | Citrus cinensis | USA | KC357553 | KC357522 | KC357456 | MF418280 | KC357488 |
| CBS 135422 | Citrus sp. | USA | KC843311 | KC843071 | KC843187 | MF418281 | KC843157 | |
| D. eres | CPC 29331 | Vitis vinifera | France | MG281034 | MG281555 | MG281207 | KC343631 | KC343389 |
| CBS 138594 | Ulmus laevis | Germany | KJ210529 | KJ210550 | KJ420799 | KC343637 | KC343395 | |
| CBS 143344 | Vitis vinifera | Czech Republic | MG281020 | MG281541 | MG281193 | MG281366 | MG281715 | |
| CAA756 | Banksia sp. | Portugal | MW040531 | MW052385 | MW091320 | MW052384 | MW091319 | |
| CAA954 | Pinus pinaster | Portugal | MN190309 | MT309431 | MT309457 | MT309440 | MT309448 | |
| CAA1001 | Quercus suber | Portugal | MT237172 | MT309432 | MT309458 | MT309441 | MT309449 | |
| D. eres (syn. D. alnea) | CBS 146.46 | Alnus sp. | The Netherlands | KC343008 | KC343734 | KC343976 | KC343492 | KC343250 |
| CBS 159.47 | Alnus sp. | The Netherlands | KC343009 | KC343735 | KC343977 | KC343493 | KC343251 | |
| D. eres (syn. D. alleghaniensis) | CBS 495.72 | Betula alleghaniensis | Canada | FJ889444 | GQ250298 | KC843228 | KC343491 | KC343249 |
| D. eres (syn. D. betulae) | CFCC 50469 | Betula platyphylla | China | KT732950 | KT733016 | KT733020 | KT732999 | KT732997 |
| CFCC 50470 | Betula platyphylla | China | KT732951 | KT733017 | KT733021 | KT733000 | KT732998 | |
| D. eres (syn. D. betulina) | CFCC 52562 | Betula platyphylla | China | MH121497 | MH121539 | MH121579 | MH121457 | MH121421 |
| CFCC 52561 | Betula platyphylla | China | MH121496 | MH121538 | MH121578 | MH121456 | MH121421 | |
| D. eres (syn. D. bicincta) | CBS 121004 | Juglans sp. | USA | KC343134 | KC343860 | KC344102 | KC343618 | KC343376 |
| D. eres (syn. D. biguttusis) | CGMCC 3.17081 | Lithocarpus glabra | China | KF576282 | KF576257 | KF576307 | - | - |
| D. eres (syn. D. brevicancria) | CBS 146962 | Picea pungens | USA | MN136180 | MN136153 | MN136190 | MN136178 | MN136129 |
| MIFCC 305 | Picea glauca | USA | MN136184 | MN136151 | MN136188 | MN136176 | MN136127 | |
| D. eres (syn. Diaporthe cf. nobilis) | CBS 124030 | Malus pumila | New Zealand | KC343149 | KC343875 | KC344117 | KC343633 | KC343391 |
| CBS 134470 | Castanea sativa | Australia | KC343146 | KC343872 | KC344114 | KC343630 | KC343388 | |
| CBS 587.79 | Pinus pentaphylla | Japan | KC343153 | KC343879 | KC344121 | KC343637 | KC343395 | |
| D. eres (syn. D. camptothecicola) | CFCC 51632 | Camptotheca acuminata | China | KY203726 | KY228887 | KY228893 | KY228881 | KY228877 |
| CFCC 51633 | Camptotheca acuminata | China | KY203727 | KY228888 | KY228894 | KY228882 | KY228878 | |
| D. eres (syn. D. castaneae-mollissimae) | DNP128 | Vaccinium corymbosum | China | KC763096 | KJ210561 | KJ420801 | KJ420852 | KJ435040 |
| D. eres (syn. D. celeris) | CBS 143349 | Vitis vinifera | UK | MG281017 | MG281538 | MG281190 | MG281363 | MG281712 |
| CBS 143350 | Vitis vinifera | UK | MG281018 | MG281539 | MG281191 | MG281364 | MG281713 | |
| D. eres (syn. D. celastrina) | CBS 139.27 | Celastrus sp. | USA | KC343047 | KC343773 | KC344015 | KC343531 | KC343289 |
| D. eres (syn. D. chensiensis) | CFCC 52567 | Abies chensiensis | China | MH121502 | MH121544 | MH121584 | MH121462 | MH121426 |
| CFCC 52568 | Abies chensiensis | China | MH121503 | MH121545 | MH121585 | MH121463 | MH121427 | |
| D. eres (syn. D. cotoneastri) | CBS 135428 | Juglans cinerea | USA | KC843328 | KC843121 | KC843229 | KC343630 | KC343388 |
| CBS 439.82 | Cotoneaster sp. | UK | KC343090 | KC343816 | KC344058 | KC343574 | KC343332 | |
| D. eres (syn. D. ellipicola) | CGMCC 3.17084 | Lithocarpus glabra | China | KF576270 | KF576245 | KF576291 | - | - |
| D. eres (syn. D. fukushii) | CBS 116953 | Pyrus pyrifolia | New Zealand | KC343147 | KC343873 | KC344115 | KC343631 | KC343389 |
| CBS 116954 | Pyrus pyrifolia | Japan | JQ807469 | JQ807418 | KJ420819 | KJ420868 | KJ435023 | |
| D. eres (syn. D. helicis) | CBS 138596 | Hedera helix | France | KJ210538 | KJ210559 | KJ420828 | KJ420875 | KJ435043 |
| D. eres (syn. D. longicicola) | CGMCC 3.17089 | Lithocarpus glabra | China | KF576267 | KF576242 | KF576291 | - | - |
| CGMCC 3.17090 | Lithocarpus glabra | China | KF576268 | KF576243 | KF576292 | - | - | |
| D. eres (syn. D. mahothocarpus) | CGMCC 3.15181 | Lithocarpus glabra | China | KC153096 | KC153087 | KF576312 | - | - |
| D. eres (syn. D. maritima) | DAOMC 250563 | Picea rubens | Canada | KU552027 | KU552022 | KU574616 | - | - |
| D. eres (syn. D. momicola) | MFLUCC 16-0113 | Prunus persica | China | KU557563 | KU557631 | KU557587 | - | KU557611 |
| MFLUCC 16-0114 | Prunus persica | China | KU557564 | KU557632 | KU557588 | - | KU557612 | |
| D. eres (syn. D. neilliae) | CBS 144.27 | Sapiraea sp. | USA | KC343144 | KC343870 | KC344112 | KC343628 | KC343386 |
| D. eres (syn. D. padina) | CFCC 52590 | Prunus padus | China | MH121525 | MH121567 | MH121604 | MH121483 | MH121443 |
| CFCC 52591 | Malus domestica | China | MH121526 | MH121568 | MH121605 | MH121484 | MH121444 | |
| D. eres (syn. D. phragmitis) | CBS 138897 | Phragmites australis | China | KP004445 | - | KP004507 | KP004503 | - |
| D. eres (syn. D. pulla) | CBS 338.89 | Hedera helix | Yugoslavia | KC343152 | KC343878 | KC344120 | KC343636 | KC343394 |
| D. eres (syn. D. rosicola) | MFLU 17-0646 | Rosa sp. | UK | MG828895 | MG829270 | MG843877 | - | MG829274 |
| D. eres (syn. D. vaccinii) | CBS 160.32 | Vaccinium macrocarpon | USA | AF317578 | GQ250326 | KC344196 | KC343712 | KC343470 |
| CBS 135436 | Vaccinium corymbosum | USA | AF317570 | JQ807380 | KC843225 | KJ420877 | KC849457 | |
| D. eres (syn. D. vacuae) | CAA829 | Vaccinium corymbosum | Portugal | MK792306 | MK828077 | MK837928 | MK871446 | MK883832 |
| MUM 19.31 | Vaccinium corymbosum | Portugal | MK792309 | MK828080 | MK837931 | MK871449 | MK883834 | |
| D. foeniculina | CBS 123208 | Foeniculum vulgare | Portugal | KC343104 | KC343830 | KC344072 | KC343588 | KC343346 |
| CBS 111553 | Foeniculum vulgare | Spain | KC343101 | KC343827 | KC344069 | KC343585 | KC343343 | |
| D. malorum | CAA740 | Malus domestica | Portugal | KY435642 | KY435629 | KY435670 | KY435650 | KY435660 |
| CBS 142383 | Malus domestica | Portugal | KY435638 | KY435627 | KY435668 | KY435648 | KY435658 | |
| D. sennicola | CFCC 51634 | Senna bicapsularis | China | KY203722 | KY228883 | KY228889 | KY228879 | KY228873 |
| CFCC 51635 | Senna bicapsularis | China | KY203723 | KY228883 | KY228889 | KY228879 | KY228873 | |
1Acronyms of culture collection: CAA—Personal Culture Collection Artur Alves, University of Aveiro, Aveiro, Portugal; CBS—Westerdijk Fungal Biodiversity Institute, Utrecht, The Netherlands; CFCC—China Forestry Culture Collection Center, Beijing, China; CGMCC—China General Microbiological Culture Collection Center, Beijing, China; CMW—Forestry and Agricultural Biotechnology Institute, University of Pretoria, South Africa; CPC—Personal Culture Collection, Pedro Crous, housed at CBS; DAOMC—The Canadian Collection of Fungal Cultures, Canada; DNP—Isolates in SMML culture collection, USDA-ARS, Beltsville, USA; MFLU—Herbarium of Mae Fah Luang University, Thailand; MIFCC—Michigan Isolate Fungal Culture Collection, Michigan, USA; MUM—Culture Collection from Micoteca da Universidade do Minho, Center for Biological Engineering of University of Minho, Braga, Portugal. Ex-type isolates are in bold. The new sequences generated in this study are in italics.
Table 2.
Alignment properties and nucleotide substitution models used for phylogenetic analyses.
| Character Status Summary | Loci and Combined Alignments | ||||||
|---|---|---|---|---|---|---|---|
| ITS | TEF1-α | TUB2 | HIS3 | CAL | 4 loci | 5 loci | |
| Total characters | 537 | 387 | 430 | 488 | 522 | 1827 | 2364 |
| Invariable characters | 434 | 183 | 273 | 342 | 307 | 1104 | 1539 |
| Informative characters (%) | 92 (17%) | 184 (47%) | 143 (33%) | 116 (23%) | 200 (38%) | 649 (35%) | 735 (31%) |
| Uninformative characters | 11 | 20 | 14 | 30 | 15 | 74 | 90 |
| Tree length (TL) | 206 | 366 | 255 | 280 | 356 | 1358 | 1647 |
| Consistency index (CI) | 0.6359 | 0.7486 | 0.7686 | 0.7036 | 0.8090 | 0.7018 | 0.6594 |
| Homoplasy index (HI) | 0.3641 | 0.2514 | 0.2314 | 0.2964 | 0.1910 | 0.2982 | 0.3406 |
| Retention index (RI) | 0.8727 | 0.8892 | 0.8959 | 0.8683 | 0.9037 | 0.8554 | 0.8311 |
| Rescaled consistency index (RC) | 0.5549 | 0.6657 | 0.6886 | 0.6109 | 0.7311 | 0.6003 | 0.5480 |
| Nucleotide substitution models 1 | K2 + G | HKY | K2 + G | KKY + G | T92 + G | TN93 + G | TN93 + G |
1 K2: Kimura 2-parameter model; T92: Tamura 3-parameter model; HKY: Hasegawa–Kishono–Yano model; TN93: Tamura-Nei parameter model; G: models of evolution assuming a gamma distribution.
Table 3.
Polymorphism and genetic diversity data for the Diaporthe eres species complex.
| Loci | Number of Haplotypes (h) | Polymorphic Sites (S) | Haplotype Diversity (hd) | Nucleotide Diversity (π) | Tajima’s D Test |
|---|---|---|---|---|---|
| ITS | 18 | 35 | 0.948 | 0.025 | 0.6097 |
| TEF1-α | 19 | 43 | 0.966 | 0.019 | −1.6281 |
| TUB2 | 17 | 43 | 0.943 | 0.022 | −0.7208 |
| HIS3 | 13 | 44 | 0.882 | 0.021 | −1.0077 |
| CAL | 12 | 13 | 0.921 | 0.009 | −0.3513 |
| 5 loci 1 | 25 | 178 | 0.990 | 0.019 | −0.7359 |
| 4 loci 2 | 24 | 143 | 0.988 | 0.018 | −1.0881 |
1 Combined alignment based on ITS, TEF1-α, TUB, HIS3 and CAL loci. 2 Combined alignments based on TEF1-α, TUB2, HIS3 and CAL loci. Note: all Tajima’s D tests resulted in values that were not statistically significant.
Table 4.
Synopsis of morphological characteristics of Diaporthe eres synonymous discussed in this study. (Note: the taxonomic description of D. neilliae was not included as its asexual morph is unknown).
Table 4.
Synopsis of morphological characteristics of Diaporthe eres synonymous discussed in this study. (Note: the taxonomic description of D. neilliae was not included as its asexual morph is unknown).
| Species | Conidiomata | Conidiophores and Conidiogenous Cells | Alpha Conidia (µm) | Beta Conidia (µm) | References |
|---|---|---|---|---|---|
| D. alleghaniensis Arnold 1967 * | Conidiomata small, with a conical shape, 200–250 µm diameter | Conidiophores 9–15 × 1–2 μm, hyaline, unbranched, ampulliform, cylindrical to sub-cylindrical. Conidiogenous phiailidic, cylindrical, slightly tapering towards apex. | Alpha conidia 7–9 × 3–4 μm, aseptate, hyaline, smooth, ovate to ellipsoidal, biguttulate or multiguttulate, base sub-truncate. | Not observed | [23] |
| D. alnea Fuckel 1870 * =Phomopsis alnea | Pycnidia with 100–200 μm diameter, globose to subglobose, embedded in tissue, erumpent at maturity | Conidiophores 9–16 × 1–2 μm, hyaline, unbranched, ampulliform, cylindrical to sub-cylindrical. Conidiogenous cells phiailidic, cylindrical, tapering towards the apex. | Alpha conidia 8–10 × 2–3 μm, aseptate, hyaline, smooth, ellipsoidal, biguttulate or multiguttulate, base subtruncate. | Not observed | [23] |
| D. betulae Tian and Fan 2016 * | Conidiomatal stromata immersed, erumpent, separate, conical, with a single locule | Conidiophores reduced to phiailidic conidiogenous cells hyaline, straight or slightly curved. | Alpha conidia hyaline, ellipsoidal, aseptate, smooth, biguttulate, 8.5–11.5 × 3.5–4.5 μm. | Not observed | [55] |
| D. betulina Tian and Yang 2018 * | Conidiomata pycnidial, conical, immersed and erumpent through the bark surface, 290–645 μm diameter | Conidiophores 12.5–17.5 × 1.5–2 μm, cylindrical, hyaline, phiailidic, branched, straight or slightly curved. | Alpha conidia hyaline, aseptate, ellipsoidal to fusiform, biguttulate, acute at both ends, 8–10 × 2.5–3 μm. | Beta conidia hyaline, aseptate, filiform, straight, eguttulate, tapering towards one apex, 26–32.5 × 1 µm | [18] |
| D. bicincta Cooke and Peck 1882 * | Pycnidia with 200–300 μm diameter, globose, erumpent at maturity, conidial cirrhus extruding from ostiole | Conidiophores 7–12 × 1–2 μm, hyaline, smooth, unbranched, cylindrical to sub-cylindrical. Conidiogenous cells 0.5–1 μm diameter, phiailidic, cylindrical, tapering towards the apex. | Alpha conidia 9–12 × 2–3.5 μm, aseptate, hyaline, ovate to ellipsoidal, biguttulate or multiguttulate, base subtruncate. | Not observed | [23] |
| D. biguttusis Gao and Cai 2015 * | Pycnidial conidiomata, dark brown, globose clustered, 79–227 μm diameter | Conidiophores 11.5–27.1 × 1.4–2.3 μm, cylindrical, single to multi-septate, densely aggregated, slightly tapering towards the apex. | Alpha conidia hyaline, biguttulate, fusiform or oval, with both ends obtuse, 5.9–8.5 × 1.9–2.6 μm. | Beta conidia hyaline, aseptate, filiform, hamate, eguttulate, tapering towards both ends, 28.1–37.9 × 1.3–2.0 μm | [56] |
| D. brevicancria Sakalidis and Medina-Mora 2021 * | Pycnidia dark brown to black, emersed in host tissue, solitary or aggregated, often with creamy yellow conidial cirrhus, 236–368 μm diameter | Conidiophores hyaline, reduced to conidiogenous cells phiailidic, and narrowing towards the apex, 7.1–17.5 × 1.0–2.4 μm. | Alpha conidia, hyaline, aseptate, oblong to ellipsoid, often biguttulate. with a sub-truncated base, 4.4–8.6 × 1.3–3.3 µm. | Beta conidia aseptate, hyaline, smooth, mostly convex at one end, hooked, 12.4–27.4 × 0.9–2.1 µm | [57] |
| D. camptothecicola Tiang and Yang 2017 | Conidiomatal pycnidia immersed or slightly erumpent through bark surface, sparse, globose to ovoid, with 560 μm diameter | Conidiophores (8.3–)12.5–15.8(−17.0) × 0.9–1.2 μm hyaline, unbranched, smooth, cylindrical, straight or slightly curved, conidiogenous cells enteroblastic, phiailidic. | Alpha conidia hyaline, aseptate, oblong, biguttulate, (4.6–)5.5–7.0(−7.5) × 1.5–1.8 μm. | Beta conidia hyaline, aseptate, filiform with obtuse ends, 19.5–28.3 × 1.0 μm | [58] |
| D. celastrina Ellis and Barthol 1902 * | Pycnidia with 200–300 μm diameter, globose, embedded in tissue, erumpent at maturity conidial cirrhus extruding from ostiole | Conidiophores 7–21 × 1–2 μm, hyaline, smooth, unbranched, ampulliform, cylindrical. Conidiogenous cells 0.5–1 μm diam, phiailidic, cylindrical, terminal, slightly tapering towards apex. | Alpha conidia 9–12 × 2–3.5 μm, aseptate, hyaline, ellipsoidal, biguttulate, multiguttulate, or eguttulate, base subtruncate. | Not observed | [23] |
| D. celeris Guarnaccia, Woodhall and Crous 2018 * | Conidiomata pycnidial, globose or irregular, solitary, erumpent, dark brown to black, 350–650 μm diameter, with yellowish translucent to brown conidial cirrhus | Conidiophores hyaline, smooth, unbranched, cylindrical, straight, 5–18 × 1–3 μm. Conidiogenous cells phiailidic, hyaline, cylindrical, 5–8 × 1–2 μm, tapering towards the apex. | Alpha conidia, aseptate, fusiform, hyaline, mono- to biguttulate and acutely rounded at both ends, 5.5–7.5 × 2–3 μm. | Beta conidia hyaline, eguttulate, filiform, curved, tapering towards both ends, 16–22.5 × 1–2 μm | [17] |
| D. chensiensis Tian and Yang 2018 * | Conidiomata pycnidial, immersed in bark, slightly erumpent discoid, ostiolate, 200–325 μm diameter | Conidiophores 8.5–13 × 2–3 μm, cylindrical, hyaline, phiailidic, unbranched, straight or slightly curved, tapering towards the apex. | Alpha conidia hyaline, aseptate, smooth, ellipsoidal, biguttulate, rounded at both ends, 6.5–11 × 2–2.2 μm. | Beta conidia present on the host only, hyaline, eguttulate, smooth, filiform, 21–28.5 × 0.8–1.1 μm | [18] |
| D. ellipicola Gao and Cai 2015 | Pycnidial conidiomata, globose, 141–338 µm diameter, erumpent, single or clustered, extruding yellowish translucent conidial droplets from the ostioles | Conidiophores cylindrical, branched, septate, hyaline, 12–22.4 × 1.1–2 µm, phiailidic, cylindrical, straight, slightly tapering towards the apex. | Alpha conidia 6–8.7 × 2–3 µm, aseptate, hyaline, smooth, biguttulate, oval, ellipsoid rounded at both ends. | Beta conidia 23.4–35.5 × 1.4–2 µm, hyaline, curved | [56] |
| D. eres Nitschke 1870 =D. castaneae-mollisimae =D. cotoneastri =D. fukushi | Pycnidia with 200–250 μm diameter, globose, embedded in tissue, erumpent at maturity, often with yellowish, conidial cirrhus extruding from ostiole | Conidiophores 10–15 × 2–3 μm, hyaline, smooth, unbranched, ampulliform, straight to sinuous. Conidiogenous cells 0.5–1 μm diameter, phiailidic, cylindrical, slightly tapering towards the apex. | Alpha conidia (6–)6.5–8.5(−9) × 3–4 μm, aseptate, hyaline, smooth, ovate to ellipsoidal, often biguttulate, base subtruncate. | Beta conidia 18–29 × 1–1.5 μm, aseptate, hyaline, smooth, fusiform to hooked, base sub-truncate | [23] |
| D. helicis Niessl 1876 * =D. nitschkei | Pycnidia with 200–300 μm diameter, globose, embedded in tissue, erumpent at maturity, often with white conidial cirrhus extruding from ostiole | Conidiophores (6–)8–15 (16.5) × 1–2 μm, hyaline, smooth, unbranched, ampulliform, cylindrical to clavate. Conidiogenous cells phiailidic, cylindrical, tapering towards the apex. | Alpha conidia (5.5–)6–8(9.5) × 2.5–3.5 μm, aseptate, hyaline, smooth, cylindrical to ellipsoidal, biguttulate or multiguttulate, base subtruncate. | Not observed | [23] |
| D. longicicola Gao and Cai 2015 | Conidiomata pycnidial, globose to subglobose, 500–750 µm diameter, dark brown to black, covered with white mycelium | Conidiophores 14.1–22.5 × 1.3–2 µm, hyaline, branched, densely aggregated, cylindrical, tapering towards the apex. | Alpha conidia 5.3–10.4 × 1.5–3.1 µm, with two big guttulate or 2–3 small guttulate, hyaline, ellipsoid or clavate, with one end obtuse and the other end acute and elongate. | Beta conidia filiform, hyaline, hamate or curved, aseptate, 25–32.2 × 1.2–2 µm | [56] |
| D. mahothocarpus Gao and Cai 2015 =Phomopsis mahothocarpus | Conidiomata globose, 200–350 μm diameter, ostiolate, deeply embedded in culture, aggregated in clusters | Conidiophores 15.5–21.8 × 1.6–2.2 μm, cylindrical, hyaline, branched, septate, straight or slightly curved. | Alpha conidia 5.5–8.0 × 1.8–2.9 μm, hyaline, aseptate, oval or fusiform, usually with one guttule at each end. | Beta conidia 21.1–28.5 × 1.2–1.9 μm, aseptate, filiform, hyaline, curved, eguttulate, with obtuse ends | [59] |
| D. maritima Tanney 2016 * | Conidiomata pycnidial, globose to subglobose, unilocular/multilocular, aggregated, dark brown to black, ostiolate, up to 300 μm diameter, with yellowish conidial mass | Conidiogenous cells phiailidic, subcylindrical to ampulliform, straight to sinuous, cylindrical or slightly tapering towards the apex, (8.5–)9–12.5(–16) × 2–3 μm. | Alpha conidia aseptate, hyaline, smooth, oblong to fusiform or ellipsoidal, apex rounded, base subtruncate, bi- to multiguttulate (10–)11–12.5(–13.5) × (3–)3.5–4 μm. | Beta conidia aseptate, hyaline, smooth, straight to hamiform or uncinate 29–40 × 1–2 μm | [60] |
| D. momicola Dissanayake, Li and Hyde 2017 | Conidiomata up to 350 μm diameter, solitary or in groups with black cylindrical ostiolate necks, subglobose | Conidiophores reduced to conidiogenous cells. | Alpha conidia 6.5–9.5 × 1.5–2 μm, hyaline, smooth, biguttulate, fusiform to oval, tapered at both ends, cylindrical to ellipsoidal. | Beta conidia 20–32 × 1–1.5 μm, scattered among the alpha conidia | [61] |
| D. nobilis Saccardo and Spegazzini 1878 | Conidiomata pycnidial, scattered to confluent, uniloculate, dark brown to black, broadly spherical to flattened, 650–700 μm high and 400–500 μm wide | Conidiophores thin walled, brown, vertically aligned, multicellular, 2–6 μm wide, elongate. Conidiogenous cells formed at the apex of the conidiophores cylindric, straight or curved. | Alpha conidia 7–9 × 3–5 μm, aseptate, cylindrical or ellipsoidal, obtuse at both ends, hyaline, generally biguttulate. | Beta conidia 20–30 × 0.3–0.8 μm, filiform, blunt at one end, pointed and usually curved at the other, hyaline, one-celled | [62] |
| D. padina Tian and Yang 2017 * | Conidiomata pycnidial, immersed in bark, scattered, slightly erumpent, light brown, one ostiole, 330–520 μm diameter | Conidiophores 5.5–12.5 × 1–1.5 μm, hyaline, unbranched, cylindrical, straight or slightly curved. | Alpha conidia hyaline, aseptate, ellipsoidal to fusiform, eguttulate, 7–8 × 1.5–2 μm. | Beta conidia hyaline, filiform, straight or hamate, eguttulate, base truncate, 21–24 × 1 µm | [18] |
| D. phragmitis Crous 2014 * | Conidiomata pycnidial, globose, up to 250 µm diameter, black, erumpent, exuding creamy conidial droplets from central ostioles | Conidiophores hyaline, smooth, septate, rarely branched, densely aggregated, cylindrical, 20–30 × 3–4 µm. Conidiogenous cells phiailidic, cylindrical, terminal, intercalary. | Alpha conidia aseptate, hyaline, smooth, multi- or bi-guttulate, fusoid to ellipsoid, tapering towards both ends, base subtruncate, 6–8.5 × 2–3 µm. | Not observed | [63] |
| D. pulla Nitschke 1870 * =Phoma pulla =Phomopsis pulla | Pycnidia with 200–300 μm diameter, globose, embedded in tissue, erumpent at maturity, black stromata, with bright yellow conidial cirrhus | Conidiophores 10–25 × 1–2 μm, hyaline, unbranched, cylindrical to clavate. Conidiogenous cells phiailidic, cylindrical, slightly tapering towards the apex. | Alpha conidia (6–)6.5–7.5 (8) × (2–) 2.5–3.5(−4) μm, aseptate, hyaline, smooth, cylindrical to ellipsoidal, biguttulate or multi-guttulate, base subtruncate. | Not observed | [23] |
| D. rosicola Wanasinghe, Jones and Hyde 2018 * | Conidiomata pycnidial, 120–160 µm diameter, solitary, semi-immersed, unilocular, globose, dark brown, ostiolate | Conidiophores hyaline, smooth, unbranched, cylindrical, straight to sinuous. Conidiogenous cells phiailidic, cylindrical, slightly tapering towards the apex. | Alpha conidia 7–9.5 × 2.4–3 µm, hyaline, biguttulate, fusiform or oval, both ends, obtuse. | Beta conidia 12–22 × 1.2–1.6 µm, hyaline, aseptate, filiform, tapering towards both ends | [64] |
| D. vaccinii Shear 1931 * =Phomopsis vaccinii | Conidiomata superficial, scattered, black, spherical to irregular, uniloculate, with ostiole circular, exuding white to yellowish cirrhus | Conidiogenous cells enteroblastic, phiailidic, with conidiophores short, 1–2 septa or multiseptate, branched. | Alpha conidia 5.9–11.3 × 2.1–3.9 μm, hyaline, fusiform, straight, guttulate, aseptate. | Beta conidia hyaline, filiform, straight or curved, eguttulate, aseptate | [65] |
| D. vacuae Hilário, Santos and Alves 2020 * | Pycnidial conidiomata, brown to black, broadly spherical, covered in white mycelium, with yellowish conidial cirrhus extruding from ostiole | Conidiophores reduced to conidiogenous cells, hyaline, smooth and straight to sinuous, broadening in the base, slightly tapering toward the apex (10.9 ± 2.2 × 1.8 ± 0.3) μm. | Alpha conidia infrequent, hyaline, smooth, cylindrical, 9.3 ± 1.1 × 2.6 ± 0.3 μm. | Beta conidia hyaline, 1-celled, smooth, filiform, frequently hooked in apical part, apex acute, 27.4 ± 2.3 × 1.6 ± 0.2 μm | [29] |
The newly synonymous introduced in the present study are marked with an asterisk (*).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hilário, S.; Gonçalves, M.F.M.; Alves, A. Using Genealogical Concordance and Coalescent-Based Species Delimitation to Assess Species Boundaries in the Diaporthe eres Complex. J. Fungi 2021, 7, 507. https://doi.org/10.3390/jof7070507
AMA Style
Hilário S, Gonçalves MFM, Alves A. Using Genealogical Concordance and Coalescent-Based Species Delimitation to Assess Species Boundaries in the Diaporthe eres Complex. Journal of Fungi. 2021; 7(7):507. https://doi.org/10.3390/jof7070507
Chicago/Turabian StyleHilário, Sandra, Micael F. M. Gonçalves, and Artur Alves. 2021. "Using Genealogical Concordance and Coalescent-Based Species Delimitation to Assess Species Boundaries in the Diaporthe eres Complex" Journal of Fungi 7, no. 7: 507. https://doi.org/10.3390/jof7070507
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.