
Providing Scale to a Known Taxonomic Unknown—At Least a 70-Fold Increase in Species Diversity in a Cosmopolitan Nominal Taxon of Lichen-Forming Fungi

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
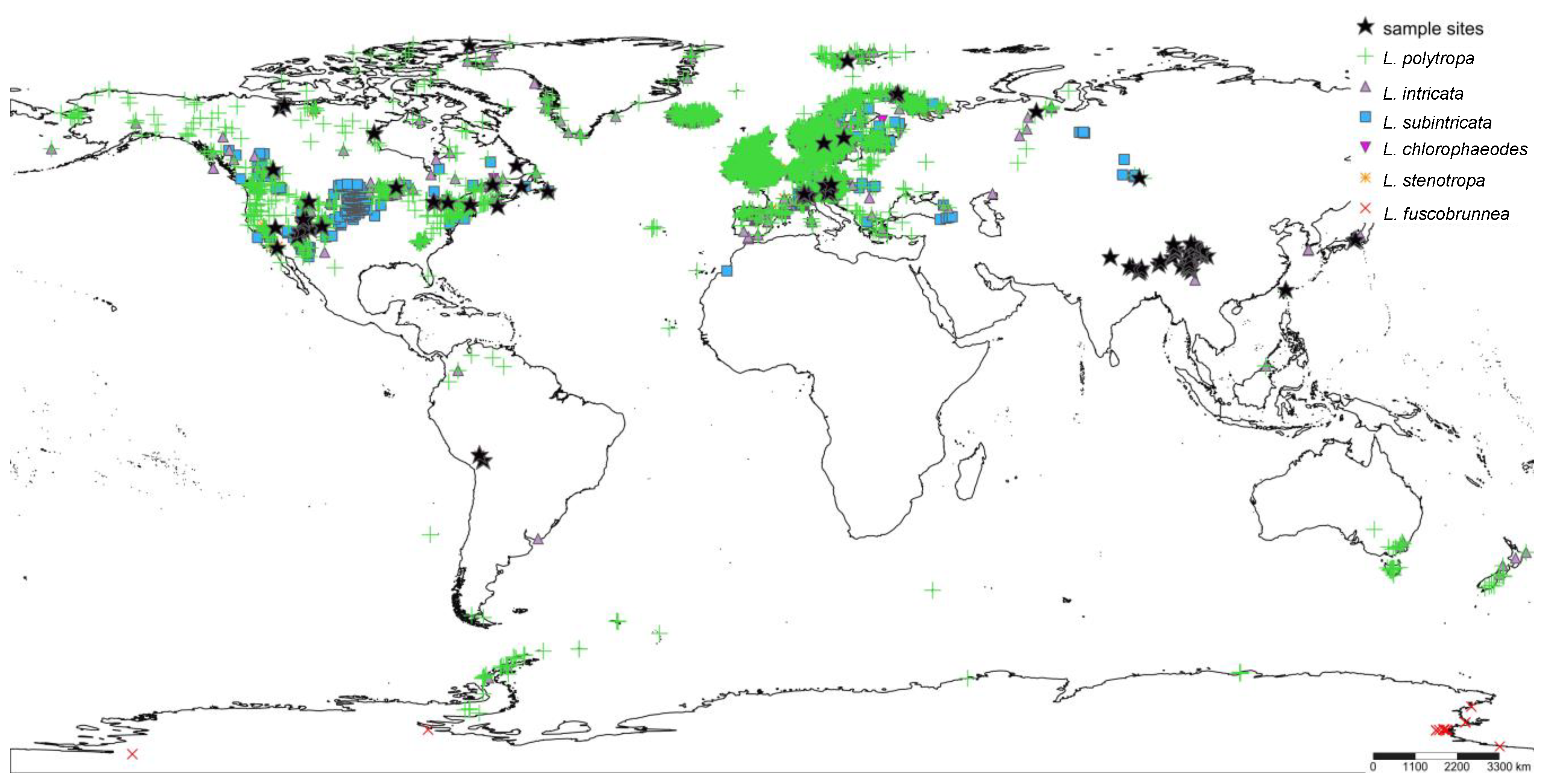
2.1. Taxon Sampling
2.2. DNA Extraction and Sequencing
2.3. Short-Read Processing and Data Assembly
2.4. Candidate Species Delimitation Using the Standard Fungal DNA Barcode
2.5. Phylogenetic Analyses and Tests of Genomic Concordance
3. Results
3.1. Sequence Data
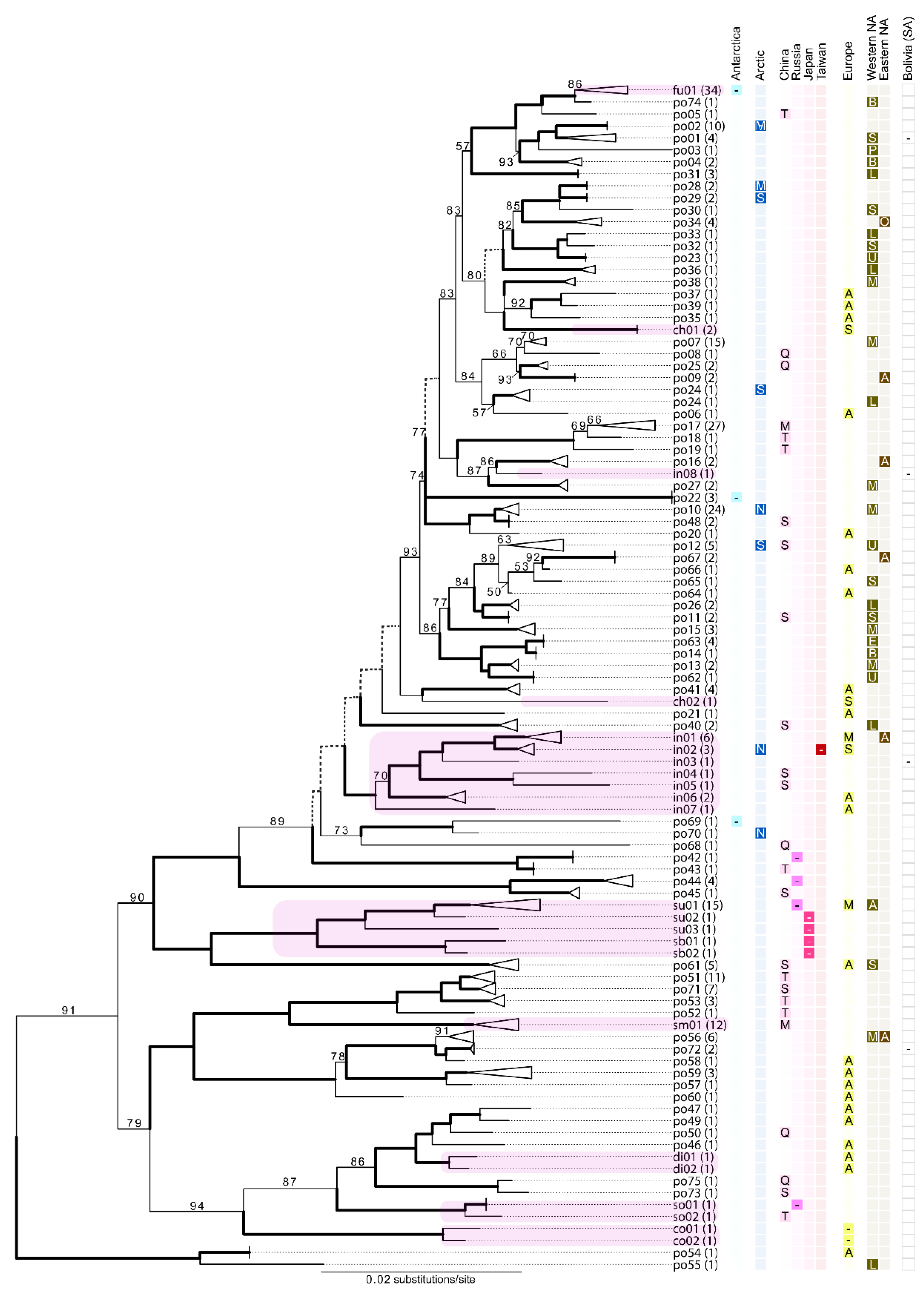
3.2. Candidate Species Inferred Using the Standard DNA Barcode (ITS)
3.3. Multi-Locus Phylogenetic Inference and Phylogenetic Concordance among Data Sets
3.4. Assessing Morphological Concordance with Species Partitions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blaxter, M.; Archibald, J.M.; Childers, A.K.; Coddington, J.A.; Crandall, K.A.; Di Palma, F.; Durbin, R.; Edwards, S.V.; Graves, J.A.M.; Hackett, K.J.; et al. Why sequence all eukaryotes? Proc. Natl. Acad. Sci. USA 2022, 119, e2115636118. [Google Scholar] [CrossRef] [PubMed]
- Ogle, W. (Ed.) Aristotle: On the Parts of Animals; Kegan Paul, Trench & Co.: London, UK, 1882. [Google Scholar]
- Burki, F.; Roger, A.J.; Brown, M.W.; Simpson, A.G. The New Tree of Eukaryotes. Trends Ecol. Evol. 2020, 35, 43–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, B.C.; Balick, M.J. Does the name really matter? The importance of botanical nomenclature and plant taxonomy in biomedical research. J. Ethnopharmacol. 2014, 152, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Borowitzka, M.A. Systematics, taxonomy and species names: Do They Matter? In The Physiology of Microalgae; Borowitzka, M.A., Beardall, J., Raven, J.A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 655–681. [Google Scholar] [CrossRef]
- Barron, E.S.; Sthultz, C.M.; Hurley, D.; Pringle, A. Names matter: Interdisciplinary research on taxonomy and nomenclature for ecosystem management. Prog. Phys. Geogr. Earth Environ. 2015, 39, 640–660. [Google Scholar] [CrossRef] [Green Version]
- Schlick-Steiner, B.C.; Steiner, F.M.; Seifert, B.; Stauffer, C.; Christian, E.; Crozier, R.H. Integrative Taxonomy: A Multisource Approach to Exploring Biodiversity. Annu. Rev. Èntomol. 2010, 55, 421–438. [Google Scholar] [CrossRef]
- Dayrat, B. Towards integrative taxonomy. Biol. J. Linn. Soc. 2005, 85, 407–415. [Google Scholar] [CrossRef]
- Smith, M.L.; Carstens, B.C. Process-based species delimitation leads to identification of more biologically relevant species. Evolution 2020, 74, 216–229. [Google Scholar] [CrossRef]
- Camargo, A.; Sites, J. Species Delimitation: A Decade After the Renaissance. In The Species Problem-Ongoing Issues; Pavlinov, I., Ed.; InTech: London, UK, 2013. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Baldrian, P.; Větrovský, T.; Lepinay, C.; Kohout, P. High-throughput sequencing view on the magnitude of global fungal diversity. Fungal Divers. 2021. [Google Scholar] [CrossRef]
- Salgado-Salazar, C.; Rossman, A.Y.; Chaverri, P. Not as Ubiquitous as We Thought: Taxonomic Crypsis, Hidden Diversity and Cryptic Speciation in the Cosmopolitan Fungus Thelonectria discophora (Nectriaceae, Hypocreales, Ascomycota). PLoS ONE 2013, 8, e76737. [Google Scholar] [CrossRef] [Green Version]
- Chethana, K.W.T.; Manawasinghe, I.S.; Hurdeal, V.G.; Bhunjun, C.S.; Appadoo, M.A.; Gentekaki, E.; Raspé, O.; Promputtha, I.; Hyde, K.D. What are fungal species and how to delineate them? Fungal Divers. 2021, 109, 1–25. [Google Scholar] [CrossRef]
- Kobmoo, N.; Mongkolsamrit, S.; Arnamnart, N.; Luangsa-Ard, J.J.; Giraud, T. Population genomics revealed cryptic species within host-specific zombie-ant fungi (Ophiocordyceps unilateralis). Mol. Phylogenet. Evol. 2019, 140, 106580. [Google Scholar] [CrossRef] [PubMed]
- Lücking, R.; Dal-Forno, M.; Sikaroodi, M.; Gillevet, P.M.; Bungartz, F.; Moncada, B.; Yánez-Ayabaca, A.; Chaves, J.L.; Coca, L.F.; Lawrey, J.D. A single macrolichen constitutes hundreds of unrecognized species. Proc. Natl. Acad. Sci. USA 2014, 111, 11091–11096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Haelewaters, D.; Schoutteten, N.; Begerow, D.; Boekhout, T.; Giachini, A.J.; Gorjón, S.P.; Gunde-Cimerman, N.; Hyde, K.D.; Kemler, M.; et al. Delimiting species in Basidiomycota: A review. Fungal Divers. 2021, 109, 181–237. [Google Scholar] [CrossRef]
- Peksa, O.; Gebouská, T.; Škvorová, Z.; Vančurová, L.; Škaloud, P. The guilds in green algal lichens—An insight into the life of terrestrial symbiotic communities. FEMS Microbiol. Ecol. 2022, 98, fiac008. [Google Scholar] [CrossRef]
- Spribille, T.; Tuovinen, V.; Resl, P.; Vanderpool, D.; Wolinski, H.; Aime, M.C.; Schneider, K.; Stabentheiner, E.; Toome-Heller, M.; Thor, G.; et al. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science 2016, 353, 488–492. [Google Scholar] [CrossRef] [Green Version]
- Mark, K.; Laanisto, L.; Bueno, C.G.; Niinemets, Ü.; Keller, C.; Scheidegger, C. Contrasting co-occurrence patterns of photobiont and cystobasidiomycete yeast associated with common epiphytic lichen species. New Phytol. 2020, 227, 1362–1375. [Google Scholar] [CrossRef]
- Rolshausen, G.; Hallman, U.; Grande, F.D.; Otte, J.; Knudsen, K.; Schmitt, I. Expanding the mutualistic niche: Parallel symbiont turnover along climatic gradients. Proc. R. Soc. B 2020, 287, 20192311. [Google Scholar] [CrossRef]
- Honegger, R. The Symbiotic Phenotype of Lichen-Forming Ascomycetes. In Fungal Associations; Hock, B., Ed.; Springer: Berlin/Heidelberg, Germany, 2001; Volume 9, pp. 165–188. [Google Scholar]
- Spribille, T. Relative symbiont input and the lichen symbiotic outcome. Curr. Opin. Plant Biol. 2018, 44, 57–63. [Google Scholar] [CrossRef]
- Armaleo, D.; Clerc, P. Lichen chimeras: DNA analysis suggests that one fungus forms two morphotypes. Exp. Mycol. 1991, 15, 1–10. [Google Scholar] [CrossRef]
- Schneider, K.; Resl, P.; Spribille, T. Escape from the cryptic species trap: Lichen evolution on both sides of a cyanobacterial acquisition event. Mol. Ecol. 2016, 25, 3453–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücking, R.; Leavitt, S.D.; Hawksworth, D.L. Species in lichen-forming fungi: Balancing between conceptual and practical considerations, and between phenotype and phylogenomics. Fungal Divers. 2021, 109, 99–154. [Google Scholar] [CrossRef]
- Gilbert, S.F.; Sapp, J.; Tauber, A.I. A Symbiotic View of Life: We Have Never Been Individuals. Q. Rev. Biol. 2012, 87, 325–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuovinen, V.; Ekman, S.; Thor, G.; Vanderpool, D.; Spribille, T.; Johannesson, H. Two Basidiomycete Fungi in the Cortex of Wolf Lichens. Curr. Biol. 2019, 29, 476–483.e5. [Google Scholar] [CrossRef] [Green Version]
- Magain, N.; Tniong, C.; Goward, T.; Niu, D.; Goffinet, B.; Sérusiaux, E.; Vitikainen, O.; Lutzoni, F.; Miadlikowska, J. Species delimitation at a global scale reveals high species richness with complex biogeography and patterns of symbiont association in Peltigera section Peltigera (lichenized Ascomycota: Lecanoromycetes). TAXON 2018, 67, 836–870. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, S.K.; Walston, R.F.; Allen, J.L. Complete, high-quality genomes from long-read metagenomic sequencing of two wolf lichen thalli reveals enigmatic genome architecture. Genomics 2020, 112, 3150–3156. [Google Scholar] [CrossRef]
- Lücking, R.; Forno, M.D.; Moncada, B.; Coca, L.F.; Vargas-Mendoza, L.Y.; Aptroot, A.; Arias, L.J.; Besal, B.; Bungartz, F.; Cabrera-Amaya, D.M.; et al. Turbo-taxonomy to assemble a megadiverse lichen genus: Seventy new species of Cora (Basidiomycota: Agaricales: Hygrophoraceae), honouring David Leslie Hawksworth’s seventieth birthday. Fungal Divers. 2017, 84, 139–207. [Google Scholar] [CrossRef]
- Lagreca, S.; Lumbsch, H.T. Three Species of Lecanora New to North America, With Notes on Other Poorly Known Lecanoroid Lichens. Bryologist 2001, 104, 204–211. [Google Scholar] [CrossRef]
- Yakovchenko, L.S.; Davydov, E.A.; Ohmura, Y.; Printzen, C. The phylogenetic position of species of Lecanora s. l. containing calycin and usnic acid, with the description of Lecanora solaris Yakovchenko & Davydov sp. nov. Lichenologist 2019, 51, 147–156. [Google Scholar] [CrossRef]
- Obermayer, W.; Poelt, J. Contributions to the Knowledge of the Lichen Flora of the Himalayas III. On Lecanora Somervellii Paulson (Lichenized Ascomycotina, Lecanoraceae). Lichenologist 1992, 24, 111–117. [Google Scholar] [CrossRef]
- Medeiros, I.D.; Mazur, E.; Miadlikowska, J.; Flakus, A.; Rodriguez-Flakus, P.; Pardo-De la Hoz, C.J.; Cieślak, E.; Śliwa, L.; Lutzoni, F. Turnover of Lecanoroid Mycobionts and Their Trebouxia Photobionts Along an Elevation Gradient in Bolivia Highlights the Role of Environment in Structuring the Lichen Symbiosis. Front. Microbiol. 2021, 12, 774839. [Google Scholar] [CrossRef] [PubMed]
- Śliwa, L.; Flakus, A. Lecanora microloba, a new saxicolous species from Poland. Lichenologist 2011, 43, 1–6. [Google Scholar] [CrossRef]
- Purvis, O.W.; Pawlik-Skowrońska, B.; Cressey, G.; Jones, G.C.; Kearsley, A.; Spratt, J. Mineral phases and element composition of the copper hyperaccumulator lichen Lecanora polytropa. Miner. Mag. 2008, 72, 607–616. [Google Scholar] [CrossRef]
- De La Rosa, I.N.; Passo, A.; Rodríguez, J.M.; Chiapella, J.O.; Messuti, M.I. A new species and new records of Lecanora (Lecanoraceae, lichenized Ascomycota) with usnic acid from the Antarctic region. Phytotaxa 2016, 261, 185–193. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium; Fungal Barcoding Consortium Author List; Bolchacova, E.; et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Leavitt, S.D.; Zhao, Z.T.; Zhang, L.L.; Arup, U.; Grube, M.; Pérez-Ortega, S.; Printzen, C.; Śliwa, L.; Kraichak, E.; et al. Towards a revised generic classification of lecanoroid lichens (Lecanoraceae, Ascomycota) based on molecular, morphological and chemical evidence. Fungal Divers. 2016, 78, 293–304. [Google Scholar] [CrossRef]
- Culberson, C.F. Improved conditions and new data for identification of lichen products by standardized thin-layer chromatographic method. J. Chromatogr. A 1972, 72, 113–125. [Google Scholar] [CrossRef]
- Orange, A.; James, P.W.; White, F.J. Microchemical Methods for the Identification of Lichens; British Lichen Society: London, UK, 2001; p. 101. [Google Scholar]
- Dupuis, J.R.; Roe, A.D.; Sperling, F.A.H. Multi-locus species delimitation in closely related animals and fungi: One marker is not enough. Mol. Ecol. 2012, 21, 4422–4436. [Google Scholar] [CrossRef]
- Carstens, B.C.; Pelletier, T.A.; Reid, N.M.; Satler, J.D. How to fail at species delimitation. Mol. Ecol. 2013, 22, 4369–4383. [Google Scholar] [CrossRef]
- Avise, J.; Ball, R. Principles of genealogical concordance in species concepts and biological taxonomy. Oxf. Surv. Evol. Biol. 1990, 7, 45–67. [Google Scholar]
- Maharachchikumbura, S.S.N.; Chen, Y.; Ariyawansa, H.A.; Hyde, K.D.; Haelewaters, D.; Perera, R.H.; Samarakoon, M.C.; Wanasinghe, D.N.; Bustamante, D.E.; Liu, J.-K.; et al. Integrative approaches for species delimitation in Ascomycota. Fungal Divers. 2021, 109, 155–179. [Google Scholar] [CrossRef]
- Grewe, F.; Lagostina, E.; Wu, H.; Printzen, C.; Lumbsch, T. Population genomic analyses of RAD sequences resolves the phylogenetic relationship of the lichen-forming fungal species Usneaantarctica and Usneaaurantiacoatra. MycoKeys 2018, 43, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Keuler, R.; Garretson, A.; Saunders, T.; Erickson, R.J.; Andre, N.S.; Grewe, F.; Smith, R.; Lumbsch, H.T.; Huang, J.-P.; Clair, L.L.S.; et al. Genome-scale data reveal the role of hybridization in lichen-forming fungi. Sci. Rep. 2020, 10, 1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol. Biol. 2019, 1962, 227–245. [Google Scholar] [CrossRef]
- Goff, S.A.; Vaughn, M.; McKay, S.; Lyons, E.; Stapleton, A.E.; Gessler, D.; Matasci, N.; Wang, L.; Hanlon, M.; Lenards, A.; et al. The iPlant Collaborative: Cyberinfrastructure for Plant Biology. Front. Plant Sci. 2011, 2, 34. [Google Scholar] [CrossRef] [Green Version]
- Merchant, N.; Lyons, E.; Goff, S.; Vaughn, M.; Ware, D.; Micklos, D.; Antin, P. The iPlant Collaborative: Cyberinfrastructure for Enabling Data to Discovery for the Life Sciences. PLoS Biol. 2016, 14, e1002342. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.G.; Gardner, E.M.; Liu, Y.; Medina, R.; Goffinet, B.; Shaw, A.J.; Zerega, N.; Wickett, N. HybPiper: Extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Appl. Plant Sci. 2016, 4, 1600016. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Bertels, F.; Silander, O.; Pachkov, M.; Rainey, P.B.; van Nimwegen, E. Automated Reconstruction of Whole-Genome Phylogenies from Short-Sequence Reads. Mol. Biol. Evol. 2014, 31, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Tonini, J.; Moore, A.; Stern, D.; Shcheglovitova, M.; Ortí, G. Concatenation and Species Tree Methods Exhibit Statistically Indistinguishable Accuracy under a Range of Simulated conditions. PLoS Curr. 2015, 7. [Google Scholar] [CrossRef]
- Rabiee, M.; Mirarab, S. SODA: Multi-locus species delimitation using quartet frequencies. Bioinformatics 2021, 36, 5623–5631. [Google Scholar] [CrossRef] [PubMed]
- Kroken, S.; Taylor, J.W. A gene genealogical approach to recognize phylogenetic species boundaries in the lichenized fungus Letharia. Mycologia 2001, 93, 38–53. [Google Scholar] [CrossRef]
- Del-Prado, R.; Buaruang, K.; Lumbsch, H.T.; Crespo, A.; Divakar, P.K. DNA sequence-based identification and barcoding of a morphologically highly plastic lichen forming fungal genus (Parmotrema, Parmeliaceae) from the tropics. Bryologist 2019, 122, 281–291. [Google Scholar] [CrossRef]
- Leavitt, S.; Fernández-Mendoza, F.; Pérez-Ortega, S.; Sohrabi, M.; Divakar, P.K.; Lumbsch, T.; Clair, L.S. DNA barcode identification of lichen-forming fungal species in the Rhizoplaca melanophthalma species-complex (Lecanorales, Lecanoraceae), including five new species. MycoKeys 2013, 7, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Divakar, P.K.; Leavitt, S.D.; Molina, M.C.; Del-Prado, R.; Lumbsch, H.T.; Crespo, A. A DNA barcoding approach for identification of hidden diversity in Parmeliaceae (Ascomycota): Parmelia sensu strictoas a case study. Bot. J. Linn. Soc. 2015, 180, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Leavitt, S.D.; Kraichak, E.; Vondrak, J.; Nelsen, M.; Sohrabi, M.; Perez-Ortega, S.; Clair, L.L.S.; Lumbsch, T. Cryptic diversity and symbiont interactions in rock-posy lichens. Mol. Phylogenet. Evol. 2016, 99, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavitt, S.D.; Johnson, L.A.; Goward, T.; Clair, L.L.S. Species delimitation in taxonomically difficult lichen-forming fungi: An example from morphologically and chemically diverse Xanthoparmelia (Parmeliaceae) in North America. Mol. Phylogenet. Evol. 2011, 60, 317–332. [Google Scholar] [CrossRef]
- Stanton, D.W.G.; Frandsen, P.; Waples, R.K.; Heller, R.; Russo, I.-R.M.; Orozco-Terwengel, P.A.; Pedersen, C.-E.T.; Siegismund, H.R.; Bruford, M.W. More grist for the mill? Species delimitation in the genomic era and its implications for conservation. Conserv. Genet. 2019, 20, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, M.; Grewe, F.; Thomas, A.; Harrison, C.H.; Lindgren, H.; Muggia, L.; Clair, L.L.S.; Lumbsch, H.T.; Leavitt, S.D. Characterizing the ribosomal tandem repeat and its utility as a DNA barcode in lichen-forming fungi. BMC Evol. Biol. 2020, 20, 2. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Mo, Z.; Yang, J.; Cai, J.; Ye, L.; Zou, J.; Qin, H.; Zheng, W.; Hollingsworth, P.M.; Li, D.; et al. Testing genome skimming for species discrimination in the large and taxonomically difficult genus Rhododendron. Mol. Ecol. Resour. 2022, 22, 404–414. [Google Scholar] [CrossRef]
- Leavitt, S.D.; Grewe, F.; Widhelm, T.; Muggia, L.; Wray, B.; Lumbsch, H.T. Resolving evolutionary relationships in lichen-forming fungi using diverse phylogenomic datasets and analytical approaches. Sci. Rep. 2016, 6, 22262. [Google Scholar] [CrossRef] [PubMed]
- Widhelm, T.J.; Grewe, F.; Goffinet, B.; Wedin, M.; Goward, T.; Coca, L.F.; Distefano, I.; Košuthová, A.; Lumbsch, H.T. Phylogenomic reconstruction addressing the Peltigeralean backbone (Lecanoromycetes, Ascomycota). Fungal Divers. 2021, 110, 59–73. [Google Scholar] [CrossRef]
- Grewe, F.; Ametrano, C.; Widhelm, T.J.; Leavitt, S.; Distefano, I.; Polyiam, W.; Pizarro, D.; Wedin, M.; Crespo, A.; Divakar, P.K.; et al. Using target enrichment sequencing to study the higher-level phylogeny of the largest lichen-forming fungi family: Parmeliaceae (Ascomycota). IMA Fungus 2020, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Leaché, A.D.; Fujita, M.K.; Minin, V.N.; Bouckaert, R.R. Species Delimitation using Genome-Wide SNP Data. Syst. Biol. 2014, 63, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, J.; Holder, M.T.; Knowles, L.L. Incorporating the speciation process into species delimitation. PLoS Comput. Biol. 2021, 17, e1008924. [Google Scholar] [CrossRef]
- Fujita, M.K.; Leaché, A.D.; Burbrink, F.T.; McGuire, J.A.; Moritz, C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 2012, 27, 480–488. [Google Scholar] [CrossRef]
- Leavitt, S.D.; Westberg, M.; Nelsen, M.; Elix, J.A.; Timdal, E.; Sohrabi, M.; Clair, L.L.S.; Williams, L.; Wedin, M.; Lumbsch, H.T. Multiple, Distinct Intercontinental Lineages but Isolation of Australian Populations in a Cosmopolitan Lichen-Forming Fungal Taxon, Psora decipiens (Psoraceae, Ascomycota). Front. Microbiol. 2018, 9, 283. [Google Scholar] [CrossRef] [Green Version]
- Lagostina, E.; Andreev, M.; Grande, F.D.; Grewe, F.; Lorenz, A.; Lumbsch, H.T.; Rozzi, R.; Ruprecht, U.; Sancho, L.G.; Søchting, U.; et al. Effects of dispersal strategy and migration history on genetic diversity and population structure of Antarctic lichens. J. Biogeogr. 2021, 48, 1635–1653. [Google Scholar] [CrossRef]
- Divakar, P.K.; Wei, X.; McCune, B.; Cubas, P.; Boluda, C.G.; Leavitt, S.D.; Crespo, A.; Tchabanenko, S.; Lumbsch, H.T.; Crespo, H.A. Parallel Miocene dispersal events explain the cosmopolitan distribution of the Hypogymnioid lichens. J. Biogeogr. 2019, 46, 945–955. [Google Scholar] [CrossRef]
- Garrido-Benavent, I.; Pérez-Ortega, S. Past, present, and future research in bipolar lichen-forming fungi and their photobionts. Am. J. Bot. 2017, 104, 1660–1674. [Google Scholar] [CrossRef] [Green Version]
- Boluda, C.; Rico, V.; Divakar, P.; Nadyeina, O.; Myllys, L.; McMullin, R.; Zamora, J.; Scheidegger, C.; Hawksworth, D. Evaluating methodologies for species delimitation: The mismatch between phenotypes and genotypes in lichenized fungi (Bryoria sect. Implexae, Parmeliaceae). Pers. Mol. Phylogeny Evol. Fungi 2019, 42, 75–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedosov, A.; Achaz, G.; Gontchar, A.; Puillandre, N. MOLD, a novel software to compile accurate and reliable DNA diagnoses for taxonomic descriptions. Mol. Ecol. Resour. 2022, in press. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Clancy, J.; Jensen, J.; McMullin, R.T.; Wang, L.; Leavitt, S.D. Providing Scale to a Known Taxonomic Unknown—At Least a 70-Fold Increase in Species Diversity in a Cosmopolitan Nominal Taxon of Lichen-Forming Fungi. J. Fungi 2022, 8, 490. https://doi.org/10.3390/jof8050490
Zhang Y, Clancy J, Jensen J, McMullin RT, Wang L, Leavitt SD. Providing Scale to a Known Taxonomic Unknown—At Least a 70-Fold Increase in Species Diversity in a Cosmopolitan Nominal Taxon of Lichen-Forming Fungi. Journal of Fungi. 2022; 8(5):490. https://doi.org/10.3390/jof8050490
Chicago/Turabian StyleZhang, Yanyun, Jeffrey Clancy, Jacob Jensen, Richard Troy McMullin, Lisong Wang, and Steven D. Leavitt. 2022. "Providing Scale to a Known Taxonomic Unknown—At Least a 70-Fold Increase in Species Diversity in a Cosmopolitan Nominal Taxon of Lichen-Forming Fungi" Journal of Fungi 8, no. 5: 490. https://doi.org/10.3390/jof8050490