
Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot



1
Innovative Institute for Plant Health, Zhongkai University of Agriculture and Engineering, Guangzhou 510225, China
2
College of Plant Protection, Fujian Agriculture and Forestry University, Fuzhou 350002, China
3
Key Laboratory of Green Prevention and Control on Fruits and Vegetables in South China, Ministry of Agriculture and Rural Affairs, Zhongkai University of Agriculture and Engineering, Guangzhou 510225, China
*
Authors to whom correspondence should be addressed.
J. Fungi 2022, 8(8), 869; https://doi.org/10.3390/jof8080869
Submission received: 28 July 2022
/
Revised: 12 August 2022
/
Accepted: 15 August 2022
/
Published: 18 August 2022
(This article belongs to the Special Issue Genomics of Fungal Plant Pathogens)
Abstract
:The Cylindrocladium black rot caused by Calonectria ilicicola is a destructive disease affecting a broad range of crops. Herein, we study virulence-associated genes of C. ilicicola Ci14017 isolated from diseased peanut roots (Arachis hypogaea L.). Ci14017 was identified via phylogenetic analysis of the internal transcribed spacer region and standard Koch’s postulate testing. Virulence-associated genes were based on genome analyses and comparative analysis of transcriptome and proteome profiles of sensitive and resistant peanut cultivars. Ci14017 identified as C. ilicicola has a 66 Mb chromosome with 18,366 predicted protein-coding genes. Overall, 46 virulence-associated genes with enhanced expression levels in the sensitive cultivars were identified. Sequence analysis indicated that the 46 gene products included two merops proteins, eight carbohydrate-active enzymes, seven cytochrome P450 enzymes, eight lipases, and 20 proteins with multi-conserved enzyme domains. The results indicate a complex infection mechanism employed by Ci14017 for causing Cylindrocladium black rot in peanuts.
1. Introduction
Cylindrocladium black rot (CBR) in peanuts is a destructive fungal disease caused by Calonectria ilicicola (anamorph: Cylindrocladium parasiticum) [1,2]. The associated black and rot symptoms develop on peanut (Arachis hypogaea L.) needles, pods, and roots, eventually leading to wilting of the whole plant [3]. CBR was first discovered in Georgia, USA in 1965 [1], and has spread to various peanut production areas, including the southeastern United States, Japan, India, and Australia [1,2,4,5]. In China, the C. ilicicola pathogen has been reported to cause disease in several host plants, including Zingiber officinale Rosc., Medicago sativa L., Manglietia deciduas, and Vaccinium spp. [6,7,8,9]. Owing to the potential threat it poses, C. ilicicola was listed in the “List of Entry Phytosanitary Pests of the People’s Republic of China” in 2007 [10].
Calonectria ilicicola is a soil-borne pathogen transmitted mainly through seeds to invade hosts, which best explains difficulties in effective prevention and control [11]. Soil fumigation with methyl isothiocyanate does suppress CBR occurrence in severely affected fields to a degree, but at too high a cost [12]. The discovery of resistance mechanisms has been difficult, as no highly resistant varieties have been introduced yet, with only some medium-resistant peanut varieties identified [13]. Therefore, understanding the detailed infection mechanisms of C. ilicicola will help explore additional approaches to control CBR disease.
Pathogenic fungi attack host plants through several strategies, utilizing adhesion to the plant surface, formation of a particular infection structure, surface breakthrough, colonization, and amplification in planta [14]. The fungi enter plant tissues through stomata or wounds via the haustorium, infection cushion, and the appressorium, a critical infection structure [15,16]. Plant fungi secrete various enzymes that act as cell wall degradation enzymes, toxins, or growth regulatory substances for optimal disease development [17,18,19]. Furthermore, fungi release a repertoire of effector proteins into host cells, helping to modify host defenses and promote susceptibility [20]. These effectors are defined, in a narrow sense, as secreted proteins of less than 300 amino acids and rich in cysteine [21]. They are further characterized by N-terminal signal peptides, no transmembrane domain, multiple repeating elements, and small-molecule proteins [22,23].
RNA and protein sequencing technologies are powerful tools for the discovery of functional fungal genes or proteins involved in virulence. For example, the genes involved in degradation enzyme activity and nucleic acid metabolism were found to be required for virulence through the comparison of transcriptome and proteome data in susceptible and resistant Spongospora subterranea hosts [24]. Although CBR has been reported in China for many years, little is known regarding the molecular pathogenic mechanisms employed by C. ilicicola. The purpose of this study was to identify candidate C. ilicicola virulence-related genes. Virulence-associated genes in C. ilicicola strain Ci14017 were screened on a susceptible peanut cultivar, P562, and a resistant cultivar, T09, via combined transcriptome and proteome analysis. The current results provide global knowledge on the putative pathogenic factors of C. ilicicola, which will add to our understanding of the genetic basis of CBR development.
2. Materials and Methods
2.1. Fungal Isolation
The pathogenic fungus was isolated from diseased peanut roots collected from Meizhou City, Guangdong Province, China. A standard tissue culture isolation method was performed on potato dextrose agar (PDA) for 7 days at 25 °C until fungus growth [25]. To purify the pathogen, six single fungal spores were transferred to a new PDA medium for hyphal growth. After 7 days of culture, the mycelia were selected to observe the micromorphological characteristics of the isolated strains under a light microscope and photographed for morphological identification [26]. After genomic sequencing and identification, the C. ilicicola strain Ci14017 was maintained on cryovials containing 25% glycerol and kept at −80 °C for long term storage and on PDA slope in 10–15 °C for short term storage. Both were stored at the Innovation Institute for Plant Health of Zhongkai University of Agriculture and Engineering.
2.2. ITS Sequencing and Phylogenetic Analysis
The genomic DNA of Ci14017 was extracted using the DNeasy Mini Kit (50, QIAGEN Hilden, Germany). The internal transcriber spacer (ITS) region was amplified via PCR using the primer pair ITS1 (5′-TTCCGAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) [27]. PCR was performed as per the following protocol: initial denaturation at 94 °C for 2 min, followed by 30 cycles at 98 °C for 10 s, 50 °C for 30 s, 72 °C for 1 min, and a final extension at 72 °C for 10 min. The 561-bp purified PCR fragment was sequenced, and sequencing data were subjected to a BLASTn homology search against the NCBI nucleotide database (http://www.ncbi.nlm.nih.gov/), accessed on 27 April 2022. ITS nucleotide sequences from the Ci14017 isolate, 16 reference Calonectria species, and an outgroup strain of Neonectria major were used to establish a phylogenetic tree based on maximum likelihood, maximum parsimony, and MrBayes analyses with RaxML v2.0 [28], PAUP v4.0 [29], and MrBayes v3.2.6 [30], respectively.
2.3. Inoculation of Peanut Plants with C. ilicicola
Seeds of the peanut-sensitive cultivar P562 were immersed in 0.5 wt% sodium hypochlorite solution for 5 min for disinfection, then rinsed with sterile water three times. Twice the weight of seeds sterile water was added, and the peanut seeds were immersed for 12 h (25 ± 1 °C, in dark), rinsed with distilled water three times, and then sprouted in moist environment Petri dishes in an artificial climate chamber for germination (25 ± 1 °C, 70–100% humidity, in dark). Normally, peanut seeds germinate in two to three days in such conditions. Germinated seeds were cultivated in plastic plots (36 cm × 25 cm × 13 cm) containing sterile nutrient water (25 ± 1 °C, light/dark:16 h/8 h). The previous stages were carried out in a sterile environment. The C. ilicicola Ci14017 strain was cultured on potato dextrose (PD) for 5 days at 25 °C, and 1 g of fungal sample was resuspended in 100 mL sterile water (106/mL conidial). Ten-day-old plants were inoculated with the conidial suspension. Control plants were inoculated with 100 mL sterile water. CBR symptoms were scored 9 days post-inoculation. To complete Koch’s postulate testing, C. ilicicola was re-isolated from diseased seedlings and subjected to ITS region analysis. Three replicates of the inoculation experiment were done.
2.4. Genome Sequencing and De Novo Assembly
Genome sequencing was carried out using an Illumina HiSeq 2000 Sequencer (Illumina/Solexa), generating 100-bp paired-end reads. Reads were de novo assembled using ABySS 1.5.2 (http://www.bcgsc.ca/platform/bioinfo/software/abyss), accessed on 18 April 2022, Velvet 1.2.1 (http://www.ebi.ac.uk/~zerbino/velvet), accessed on 21 April 2022, and SOAP denovo 2.04 (http://soap.genomics.org.cn/soapdenovo.html), accessed on 24 April 2022 with a k-mer setting of 31 to 97. The assembled genome was then assessed for completeness using BUSCO v3.0 [31] (Table S1).
2.5. Gene Prediction and Annotation
The interspersed repetitive elements of Ci14017 were determined and annotated with RepeatMasker version open-4.0.5 (http://www.repeatmasker.org), accessed on 26 April 2022, followed by rRNA and tRNA detection using RNAmmer v1.2 and tRNAscan-SE v1.3.1, respectively [32,33]. For the sequencing of coding genes, AUGUSTUS v3.3.3 (http://bioinf.uni-greifswald.de/augustus/binaries/), accessed on 27 April 2022 was used as a pretest software from the head, and Fusarium graminearum was used as a reference species for de novo gene prediction. The gene pool of BUSCO was selected from four database files of Ascomycota, fungi, Hypocreales, and Sordariomycetes. The predicted coding sequences were searched against the NCBI (http://www.ncbi.nlm.nih.gov/), accessed on 27 April 2022 and Swiss-Prot protein databases (http://www.uniprot.org/), accessed on 27 April 2022 using BLASTP, with an e-value of ≤1 × 10−5. The predicted coding sequences were analyzed using Blast2GO (http://www.geneontology.org/), accessed on 28 April 2022 for Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway mapping (http://www.genome.jp/kegg/), accessed on 28 April 2022. Functional classification of the predicted proteins was performed using KOG (http://ww.ncbi.nlm.nih.gov/KOG/), accessed on 28 April 2022, whereas fungal pathogenicity domains were determined using the MEROPS [34], DFVF [35] (http://sysbio.unl.edu/DFVF/), accessed on 28 April 2022, lipase [36], cytochrome P450 (CYP450) [37], pathogen–host interaction database (PHI-base) [38,39] (http://www.phi-base.org/downloadLink.htm), accessed on 28 April 2022, and CAZymes databases [40] (http://cricket.ornl.gov/cgi-bin/cat.cgi), accessed on 28 April 2022 via BLASTP, with an e-value ≤1 × 10−5.
Genomic collinearity analysis was performed on the whole genomes of Ci14017 and F. graminearum using MCScanX, with an e-value ≤ 1 × 10−5. The predicted genome models of Ci14017 and 18 fungal species were subjected to homologous gene analysis. A comparative analysis was performed using these data. All fungal genomes were selected based on the consanguinity strategy with the eight Calonectria species, including four isolates (C. pseudonaviculata cb12013, C. pseudonaviculata cb12034, C. canadiana cc13392, and C. leucothoes cl13394) from our laboratory and Professor Tom Hsiang’s team at the University of Guelph, Canada, which has not yet been published.
Putative secreted peptides were identified based on the presence of a signal peptide secretion signal, absence of a transmembrane domain, and localization within organelles. Secreted peptides and transmembrane domains were predicted using SignalP v4.1 [41] and TMHMM v2.0 [42] (http://www.cbs.dtu.dk/services/TMHMM/), accessed on 29 April 2022, respectively. Secreted location domains were predicted using TargetP v1.1 [43] (http://www.cbs.dtu.dk/services/TargetP/), accessed on 29 April 2022 and ProtComp v9.0 [42,43] (http://linux1.soft-berry.com/berry.phtml?topic=protcomppl&group=programs&subgroup=proloc), accessed on 29 April 2022. The potential effector proteins were characterized using the following criteria: (A) annotated in PHI-base; (B) implicated in reduced virulence, loss of pathogenicity, lethal, or hypervirulence status, as described by PHI-base; and (C) localized extracellularly (secreted protein).
2.6. RNA-Sequencing
To infect peanut plants with Ci14017, 1 g fungal sample resuspended in 100 mL sterile water (106/mL) was inoculated on 10-day-old resistant cultivar T09 and the susceptible cultivar P562. At 9 days post-inoculation, root segments from the three treated seedlings were sampled. Ci14017 samples cultured in PDA medium were used as controls to monitor gene expression changes. For each sample, a pool was generated from three repeated collections. Total RNA was extracted from three sample pools using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. To construct RNA-seq libraries, cDNA was synthesized by random hexamer primers using the short fragmented mRNA, which was purified by beads containing oligo (dT) and then fragmented using EB buffer. After amplification, the quality of the library was monitored using an Agilent Bioanalyzer 2100 system. After Illumina sequencing adaptors were ligated to the short fragments, sequencing was performed using an Illumina HiSeqTM 2000 (100 bp, paired-end). In the quality control step, sample quality and identification were determined using an Agilent Bioanalyzer 2100 system (Agilent Technologies, Inc. La Jolla, CA, USA) and an ABI Step One Plus Real-Time PCR system (Applied Biosystems, Foster, CA, USA).
Clean reads were mapped to the reference genome (this study) using Bowtie2. FPKM values (expected number of fragments per kilobase of transcript sequence per million base pairs sequenced) of each unigene were calculated using RSEM (v1.3.0). Subsequently, the FPKM values were subjected to comparative analysis between sequencing groups with GFOLD to show relative expression levels [44]. Differences with an absolute fold change of FPKM value > 2 and q value ≤ 0.001 were considered statistically significant, and these unigenes were considered differentially expressed genes (DEGs).
2.7. Proteome Analysis
Proteins were extracted from three Ci14017 sample pools, as previously described [45]. Liquid chromatography/mass spectrometry (LC/MS) was performed to identify proteins showing differential expression levels between Ci14017 sample pools. The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The tryptic peptides were dissolved in 0.1% formic acid (solvent A) and then separated using a Bruker NanoElute (Germany) ultra-high performance liquid chromatography (UPLC) system. The gradient was comprised of an increase from 6% to 22% solvent B over 70 min, 22% to 32% in 14 min and climbing to 80% in 3 min, then holding at 80% for the last 3 min, all at a constant flow rate of 300 nL/min on UPLC system. The peptides were subjected to capillary ion source followed by tandem MS in Bruker timsTOF Pro (Germany) coupled online to the UPLC. The ion source voltage applied was 1.4 kV. The m/z scan range was 100 to 1700 for secondary MS. The data acquisition mode used parallel accumulation–serial fragmentation (PASEF) mode.
The resulting MS data were processed using Maxquant search engine (v.1.6.6.0). Tandem mass spectra were searched against the Arachis hypogaea L. (data not shown) and Calonectria ilicicola database (101,330 and 8978 proteins) concatenated with a reverse decoy database. Trypsin/P was specified as a cleavage enzyme allowing up to 2 missing cleavages. The mass tolerance for precursor ions was 70 ppm in the first search and 70 ppm in the main search, and the mass tolerance for fragment ions was 0.04 Da. False discovery rate (FDR) was adjusted to <1%. Proteins with a fold change (FC) greater than two were identified as differentially expressed.
2.8. Bioinformatic Analysis of Differentially Expressed Proteins (DEPs)
Multiple sequence alignment for every DEP was conducted using MAFFT (http://www.ebi.ac.uk/Tools/msa/mafft/), accessed on 2 May 2022 [46]. RaxML was used to construct a phylogenetic tree based on the maximum likelihood method [28]. Upset analysis was performed using the OmicStudio tool (http://www.omicstudio.cn/tool), accessed on 2 May 2022. The proteins were then subjected to GO annotation and InterProScan v5.14–53.0 (http://www.ebi.ac.uk/interpro/), accessed on 5 May 2022. Pathway involvement was determined using KEGG Mapper (http://www.kegg.jp/kegg/mapper.html), accessed on 5 May 2022. Heat maps were constructed and visualized using ChiPlot (http://www.chiplot.online/), accessed on 6 May 2022. The conserved domains were predicted using the Conserved Domain Database Structure in NCBI (http://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi?), accessed on 6 May 2022.
3. Results
3.1. Identification of the C. ilicicola Ci14017 Pathogen Isolated from Peanut
The CBR pathogen Ci14017 was isolated from diseased peanut roots as per a standard tissue culture isolation method. To verify the pathogenicity of Ci14017 in peanut plants, a 1% mycelium suspension was inoculated on 10-days-old susceptible cultivar P562. At 9 days post-inoculation, disfigured spots from brown to dark brown and necrotic rot appeared in the roots, with symptoms similar to those of diseased roots collected from the field (Figure 1a). Furthermore, the pathogen Ci14017 was re-isolated from symptomatic roots inoculated with mycelium suspension (data not shown). These results demonstrated that the Ci14017 isolate was the causal agent of peanut CBR.
Molecular identification of the Ci14017 isolate was performed via PCR amplification of the ITS gene region. The obtained 561-bp ITS1/4 DNA fragment was subjected to a homology search in the NCBI database. Fifteen homologs were found in C. ilicicola, sharing over 99% sequence identity. Three of these showed the highest identity of 99.29%, including the homologs from isolate HF-YJ (GenBank: MK990098.1), isolate PM-HHL-F018-WD (GenBank: MK850211.1), and an unnamed C. ilicicola isolate (GenBank: MF785081.1). The ITS sequences of C. ilicicola Ci14017, NA-LiaoNing2018, and HF-YJ, as well as those from 18 representative Calonectria species, were used to construct a phylogram. In the generated phylogenetic tree, Neonectria major strain CBS_240.29 fell into an out-group. Phylogenetic analysis indicated that Ci14017 falls into the cluster of C. ilicicola species, within the same clade as C. ilicicola MF785081.1 (Figure 1b).
3.2. Genomic Assembly and Features of C. ilicicola Ci14017
A total of 1993 scaffolds with an average length of 33,272 bp were generated from sequencing. These included the 40 smallest scaffolds of 121 bp and the largest scaffold of 1,935,007 bp. The assembled draft genome was 66,310,315 bp, with a GC content of 48.22%. The 10 largest scaffolds comprised 14,067,989 bp, accounting for 2.9% of the total genome length (Table 1).
3.3. Genome and Gene Functional Annotation
The Ci14017 genome contained 18,366 coding genes with an average gene length of 1525 bp (Table 1). The gene density was three exons per gene, with an average of 522 bp. Ninety rRNAs and 423 tRNAs were predicted (Table 1). Functional annotation of coding genes in the Ci14017 genome was performed using the GO, KOG, and KEGG databases. Overall, 10,116 genes were annotated in the GO database, 8937 in the KOG database, and 8805 genes in the KEGG database (Figure 2a,b). Approximately 46.7% of these were categorized into five KEGG pathways: global and overview maps (2309), signal transduction (519), amino acid metabolism (478), carbohydrate metabolism (454), and transport and catabolism (351) (Figure 2c).
3.4. Comparative Genomics Analysis
Genomic collinearity analysis revealed that there were up to 31,678 genes in 1993 scaffolds of Ci14017 and seven chromosomes of F. graminearum strains, with 12,096 genes showing collinearity (38.18%). The 6052 collinear genes in the Ci14017 genome were distributed in 270 blocks, whereas the 6044 collinear genes in F. graminearum were located in 151 blocks with more than 10 collinearity gene pairs and five blocks with more than 100 collinearity gene pairs (Figure 3a).
Gene family analysis was performed using F. graminearum as a model to establish a time-sequence evolutionary tree, which included eight genomes of the same Calonectria genus, three genomes of the same Nectriaceae family, six genomes of the Sordariomycetes class, and one genome of the same phylum of Ascomycota. The results showed that Ci14017 was closest to C. canadiana cc13392. The time of differentiation was estimated at 20.18 million years ago. The number of Ci14017 gene families expanded by 1217 and contracted by 627 (Figure 3b).
The Ci14017 genome was additionally compared with the genomes of five Calonectria species, including C. leucothoes, C. naviculata, C. canadiana cc13392, C. pseudoreteaudii, and C. pseudonaviculata. Using F. graminearum as a model, a Venn analysis of the gene family revealed 8864 core protein-coding genes and 135 genes specific to the Ci14017 genome (Figure 3c).
3.5. Identification of Effectors
A comprehensive assessment strategy was used to predict secretory proteins with signal peptides, transmembrane domains, extracellular subcellular localization, and exocrine function. Among the 18,366 proteins predicted in the whole genome of Ci14017, 1992 were found to possess putative signal peptides at the N-terminal based on SignalP v 4.1 analysis. Subsequent transmembrane domain prediction using TMHMM v 2.0 revealed that 1849 proteins had at least two transmembrane domains. The subcellular localization of 1804 proteins were extracellular. After excluding 86 non-exocrine proteins, 1718 proteins were eventually identified as secreted.
The 1718 proteins harbored 836 protein sequences with cysteine content ≥6, of which 289 with multiple tandem repeats had ≥9. Effector proteins are critical elements for evaluating pathogenicity. Potential effector proteins were screened based on annotation in the PHI-base. Of the 289 proteins, 264 were annotated in the PHI database. Sixteen of these were encoded by genes whose knockdown or knockout would lead to pathogen death, 96 were encoded by hypervirulence genes (gene silencing causes reduced pathogenicity), five were effectors, and five were associated with mixed outcomes. In total, 122 proteins were selected as putative effectors in Ci14017 (Figure 4a).
According to the results of the BLAST comparison, among the 1718 secretory proteins, 253 were not found in the NR database. Among 122 candidate effector proteins, 16 had no homologous genes in the NR database. Among the 122 putative genes, 78 had a cysteine content greater than 2%, and 25 had a cysteine content greater than 4%, with the highest being 12.1%. The length of the signal peptide ranged from 14 to 37 aa, and the protein with a signal peptide length of 19 aa had the highest secretory content.
3.6. CAZyme Analysis
A total of 482 CAZymes were found among the 1718 secretory proteins and subjected to a CAZyme homology search using the CAZymes analysis toolkit (CAT). The predicted CAZyme distribution is summarized as follows, according to respective classes and gene counts: 82 auxiliary activity, 119 carbohydrated esterases, 168 glycoside hydrolases, five glycosyl transferases, 63 carbohydrate-binding modules, and 45 polysaccharide lyases (Figure 4b). The 63 genes of CBM were classified into 17 gene families; the 119 genes of CE were classified into eight gene families; the 168 genes of GH were classified into 48 gene families; GT genes were classified into four gene families; PL genes were classified into six families; and the 82 genes of AA were classified into seven families. We observed that CE10 (45) CAZymes were most abundant in Ci14017, followed by AA7 (39), CE5 (22), GH3 (19), GH43 (19), and PL1 (19). These enzymes are involved in the hydrolysis of carbohydrate and non-carbohydrate substrates (CEs and GHs) as well as in the oxidative degradation of lignin-based components of the plant cell wall (AA).
3.7. Identification of 46 Virulence-Associated Factors in C. ilicicola
The peanut T09 and P562 cultivars were simultaneously inoculated with Ci14017. When compared to the highly sensitive P562, T09 showed moderate resistance to C. ilicicola. At 9 days post-inoculation, P562 roots exhibited a severe dark brown and necrotic rot, whereas T09 symptoms were notably lesser, with some visible necrotic rots (Figure 5a). The roots of both T09 and P562 plants were collected for RNA-seq and label-free proteomic analysis through comparison with C. ilicicola samples cultured in a PDA medium.
The number of DEGs in P562 and T09 after inoculation with Ci14017 (PC/TC) was 5724, with 2464 upregulated genes and 3260 downregulated genes (fold change > 2) (Table S2). There were 536 DEPs in the PC/TC proteome, all of which were upregulated (Table S3).
A total of 7188 expressed genes were identified via transcriptomic analysis. Between these and the 536 upregulated proteins identified via proteomic analysis were 517 overlapping factors. According to proteomic data, another 788 proteins were expressed only in susceptible lines. Of these, 629 overlapped with the 7188 expressed genes. Further, there were 1146 co-differentially expressed genes/proteins between the transcriptome and proteome.
We performed homology searches using the PHI, DFVF, MEROPS, LIPABASE, Cytochrome P450, CAZymes database, and Ci14017 secreted proteins in order to identify potential virulence factors. Among the 1146 DEPs, 46 were identified as putative virulence-associated genes (Figure 5b). BLASTP comparison between 46 virulence-associated genes and the NR database showed homologous genes for all candidates.
GO enrichment analysis was conducted for the 46 virulence-associated genes, and the results revealed 804 enriched GO terms (Figure 5c). Genes were mainly enriched in the polysaccharide metabolic process (5 genes) and fungal-type cell wall organization or biogenesis (5 genes). Based on GO enrichment, it can be concluded that the infection with C. ilicicola leads to destruction of the peanut cell wall and intercellular structure, resulting in peanut cell decline and death. The 46 virulence-associated genes were enriched in 59 KEGG pathways, as determined via KEGG enrichment analysis (Figure 5d). Genes were mainly enriched in exosomes (five genes), chaperones and folding catalysts (four genes), and peptidases (four genes).
3.8. Sequence Analysis of the 46 Virulence-Associated Factors
The 46 virulence-associated genes were used to construct a phylogenetic tree using RaxML. The outer circles indicate the matching results for each database. There were two merops proteins, eight carbohydrate active enzymes, seven cytochrome P450 enzymes, eight lipases, 20 multi-annotated proteins (Lipase/P450:12; Lipase/P450/Merops:1; Lipase/P450/CAZymes/Secrete:1; CAZymes/Secrete:2; P450/Merops:1; Lipase/P450/CAZymes:1; Lipase/CAZymes/Secrete:1; and P450/CAZymes:1), and one unannotated protein (Figure 6). The gene and protein expression levels of 46 virulence-associated genes were analyzed, revealing expression levels in susceptible lines compared to resistant lines at both levels. At the proteomic level, these 46 virulence-associated genes were not expressed in resistant lines, but were highly expressed in susceptible lines (Figure 6 and Table 2).
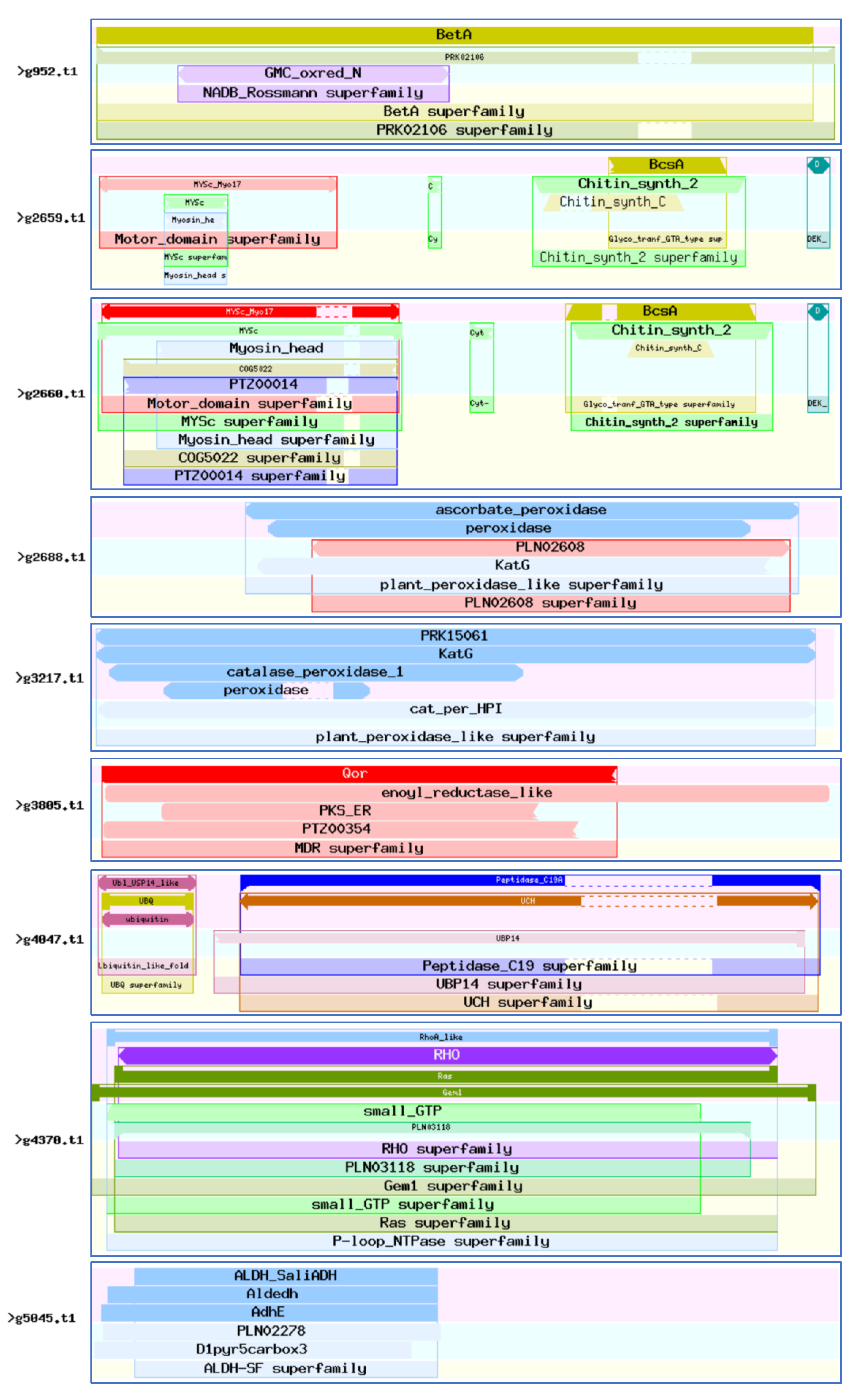
CDD (Conserved Domain Database) is a protein annotation resource consisting of a series of fully annotated domain and full-length protein sequence alignment models. The 46 virulence-associated genes were compared with the CDD database (E-value = 1 × 10−5). These genes harbored a total of 2062 CDD protein structural functional domains, 504 clusters, 111 specific features, and 19 generic features. There were six genes with more than 100 protein structural functional domains, among which g18328.t1 contained 210 protein structural functional domains and g4249.t1 contained 136 protein structural functional domains (Figure 7). These proteins are mainly related to the functions of CYP450, such as g5581.t1; ubiquitin, such as g4047.t1 and g18104.t1; chitin synthase, such as g2659.t1 and g2660.t1; fungal-specific transcription factors, such as g5899.t1; and cell division, such as g11138.t1.
4. Discussion
In this study, we isolated and characterized C. ilicicola Ci14017, which causes peanut black rot in Guangdong Province, China. To screen for candidate pathogenicity factors, we performed whole-genome sequencing with a sequencing depth of 75.84-fold. After reading 1993 scaffolds, the assembled genome size was 66,310,315 bp, coding for 18,366 genes. Based on the assessment of BUSCO profiles for the fungi core database, the matching rate of Ci14017 was up to 99%, implying a high-quality genome sequencing performance.
Effectors secreted by plant fungi play critical roles in virulence, as they modulate host defense reactions upon entry into host cells. The particular characteristics of these effectors have been summarized and successfully considered for the identification of putative effectors in a range of plant fungi. Herein, we screened for potential virulence factors according to the following criteria: annotated by PHI-base, associated with reduced virulence, loss of pathogenicity, lethal, or hypervirulence status as described by PHI-base, and localized in the extracellular space. In light of these traits, 1718 secreted proteins were annotated from the Ci14017 genome using the MEROPS, DFVF, LIPABASE, CYP450, PHI, and CAZymes databases. This demonstrates that Ci14017 harbors effectors similar to those of other known fungi.
The draft and complete genomes of Calonectria species have been continuously expanded in recent years. The number of coding genes in C. leucothoes, C. Naviculata, C. pseudoreteaudii, and C. Pseudonaviculata varied from 12,964 to 22,409. The number of coding genes in C. pseudonaviculata cb12013, C. pseudonaviculata cb12034, C. canadiana cc13392, and C. leucothoes cl13394 obtained in our laboratory in cooperation with Professor Tom Hsiang’s team from the University of Guelph, Canada, ranged from 12,979 to 17,552. Liu et al. estimated 17,308 putative protein-coding sequences in C. ilicicola F018 collected in Taiwan [47]. However, the C. ilicicola F018 genome was assembled from only 16 contigs. As determined via high-quality genome sequencing, Ci14017 contains 18,366 coding genes, a number that falls in the middle among previously reported Calonectria species. In addition to the 135 specific proteins in Ci14017, 6052 genes in Ci14017 were found to overlap with those of F. graminearum. Whole-genome evolutionary analysis revealed that Ci14017 and C. canadiana cc13392 differentiation occurred 20.18 million years ago, with Ci14017 expanding by 1217 genes and contracting by 627 genes. This suggests surprising genetic diversity among Calonectria species.
Morphology, cytology, and physiological biochemistry have been mainly described for the C. ilicicola–host interaction. This study reported the gene expression profiles of Ci14017 in planta in order to screen putative virulence-associated genes involved in CBR development. We found that 2318 genes were induced by interaction with the sensitive P562 cultivar, and 2363 genes were induced by interaction with the resistant cultivar T09. By comparing the whole-genome gene profiles of sensitive cultivar P562 and resistant cultivar T09, we observed that the expression levels of 2464 genes were higher in the sensitive cultivar P562 than in T09. Proteome analysis revealed 536 DEPs, all upregulated in the susceptible cultivar P562, implying that all of these proteins are essential for Ci14017 to cause CBR symptoms. Comparative analysis of transcriptome and proteome data revealed 46 virulence-associated genes, including those encoding ubiquitin thiolesterase, chitin synthase, cytochrome P450, and specific transcription factors.
Ubiquitin-proteasome-mediated protein degradation is an important regulatory mechanism involved in fungal cell life and host adaptation [48]. The pathway includes E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 ubiquitin ligases, which transfer ubiquitin to substrates that are then subjected to degradation in the 26S proteasome. E3 functions as the substrate recognition module of the system and is capable of transferring activated ubiquitin to its substrate. A large number of E3s in fungi are responsible for specifying degraded protein substrates [49]. Disruption of the ubiquitin-proteasome pathways in Gibberella zeae results in multiple phenotypic alterations, such as mycelial growth, sexual reproduction, and virulence [50]. The ubiquitin thiolesterase enzyme is involved in the ubiquitin-proteasome system, a cellular pathway responsible for the degradation of misfolded and damaged proteins. This emphasizes the importance of the ubiquitin-proteasome pathway for fungal virulence.
Chitin, a biopolymer composed of β-1,4-linked N-acetylglucosamine, is an important component of fungal cell walls and insect exoskeletons [51]. Chitin synthase plays an essential role in fungal cell wall stability, filamentous mycelial development, and pathogenicity [52]. In Metarhizium acridum, seven chitin synthase genes play important roles in growth and development, stress resistance, cell wall integrity, and virulence [53]. Chitin synthesis appears to play an equally essential role in Ci14017 causing disease in peanut hosts, as chitin synthases g2659.t1 and g2660.t1 of Ci14017 were specifically upregulated in the sensitive cultivar P562.
CYP450 is one of the largest heme-containing protein families and is ubiquitous in all living species [54]. CYP450 enzymes play important roles in primary and secondary metabolism as well as xenobiotic biodegradation in eukaryotes [55]. Plant pathogenic fungi possess relatively more CYP450-coding genes compared with other eukaryotes [56]. Most CYP450 enzymes in F. graminearum have redundant functions in xenobiotic detoxification and fungal development, as well as fungal reproduction and virulence [57]. Identification of the g5581.t1 (CYP450) gene as a virulence-associated gene in Ci14017 suggests the involvement of CYP450 in fungal developmental processes and virulence.
Several virulence-associated genes are enzymes necessary for metabolic processes, such as S-(hydroxymethyl) glutathione dehydrogenase (g14087.t1). To successfully colonize host plants, fungi need to acquire essential nutrients from their hosts in order to meet requirements for hyphal growth or replication. After inoculation of resistant and susceptible cultivars, the pathogen Ci14017 replicated more slowly in resistant cultivars, as these exert a suppressive effect on pathogen replication. The enhanced expression of enzymes involved in metabolic processes implies that Ci14017 replicated better in P562 than in T09. On the other hand, enhanced expression of these enzymes helped accelerate metabolic processes, providing more energy for Ci14017 replication. For example, S–(hydroxymethyl) glutathione dehydrogenase MoSFA1 mediates nitric oxide (NO) metabolism by specifically catalyzing the reduction of S–nitrosoglutathione (GSNO), which is involved in conidiation and full virulence of the rice blast fungus Magnaporthe oryzae [58].
5. Conclusions
We isolated and identified a C. ilicicola Ci14017 strain that causes CBR in peanuts. The high-quality whole genome of C. ilicicola Ci14017 was obtained by assembling 1993 scaffolds containing 18,366 coding genes. Based on the deciphered genome information, 46 virulence-associated genes were identified through comparison of the transcriptome and proteome between resistant and susceptible peanut cultivars. To our knowledge, this is the first report on virulence-associated genes in C. ilicicola, which will help us to further understand the mechanisms through which this fungus causes CBR in host plants.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/jof8080869/s1, Table S1: Comparison of whole genome assembly results of Ci14017 strains; Table S2: Ci14017 differentially expressed gene statistics (DEGs); Table S3: Ci14017 differentially expressed protein statistics (DEPs).
Author Contributions
Conceptualization, X.C. and Z.D.; methodology, X.C. and Z.D.; software, X.C. and Z.D.; validation, X.C., M.L., W.W. and Z.D.; analysis, X.C. and Z.D.; data curation, X.C. and Z.D.; writing—original draft preparation, X.C.; writing—review and editing, H.Z. and Z.D.; supervision, Z.D.; project administration, Z.D.; funding acquisition, Z.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ordinary University Innovation Project of Guangdong Province (2016KTSCX060) and Innovation and entrepreneurship training program for college students in Guangdong Province of 2020 (202011347031), awarded to Z. Dong.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw genome data of Ci14017 have been deposited in the NCBI with the accession number of SRR19913774. The raw RNA-seq data of the four peanut samples and one Ci14017 sample (hyphae) have been deposited in the NCBI with the accession number of SRR19978308, SRR19978307, SRR19977118, SRR19977117. The raw label free data of the twelve peanut samples have been deposited in the PRIDE with the accession number of PXD024578 (private data).
Acknowledgments
Thanks to Tom Hsiang of the University of Guelph, Canada, for sharing the genome-wide data of the four strains and for his guidance in genome analysis. We would like to thank Yixiong Zheng of Zhongkai University of Agriculture and Engineering for providing us with peanut seed materials during the experiment. Thanks to the Beijing Genomics Institute and Jingjie PTM BioLab for providing sequencing services.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bell, D.K.; Sobers, E.K. A peg, pod, and root necrosis of peanuts caused by a species of Calonectria. Phytopathology 1966, 56, 1361–1364. [Google Scholar]
- Wheele, T.A.; Black, M.C. First report of Cylindrocladium black rot caused by Cylindrocladium parasiticum on peanut in Texas. Plant Dis. 2005, 89, 1245. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Wang, X.J.; Feng, L.P. Progress on the research of Cylindrocladium parasiticum. China Plant Prot. 2010, 30, 19–21. [Google Scholar]
- Tazawa, J.; Takahashi, M.; Usuki, K.; Yamamoto, H. Nodulation during vegetative growth of soybean stage does not affect the susceptibility to red crown rot caused by Calonectria ilicicloa. J. Gen. Plant Pathol. 2007, 73, 180–184. [Google Scholar] [CrossRef]
- Wright, L.P.; Davis, A.J.; Wingfield, B.D.; Crous, P.W.; Brenneman, T.; Wingfiled, M.J. Population structure of Cylindrocladium parasiticum infecting peanuts (Arachis hypogaea) in Georgia, USA. Eur. J. Plant Pathol. 2010, 127, 199–206. [Google Scholar] [CrossRef]
- Pei, W.H.; Cao, J.F.; Yang, M.Y.; Zhao, Z.J.; Xue, S.M. First report of black rot of Medicago sativa caused by Cylindrocladium parasiticum (teleomorph Calonectria ilicicola) in Yunnan Province, China. Plant Dis. 2015, 99, 890. [Google Scholar] [CrossRef]
- Fei, N.Y.; Qi, Y.B.; Meng, T.T.; Fu, J.F.; Yan, X.R. First report of root rot caused by Calonectria ilicicola on blueberry in Yunnan Province, China. Plant Dis. 2018, 102, 1036. [Google Scholar] [CrossRef]
- Zhang, Q.M.; Zhou, D.M.; Jiang, W.; Zhu, H.L.; Deng, S.; Wei, L.H. First report of soft rot of ginger caused by Calonectria ilicicola in Guangxi Province, China. Plant Dis. 2019, 104, 993. [Google Scholar] [CrossRef]
- Yi, R.H.; Su, J.J.; Li, H.J.; Li, D.; Long, G.G. First report of root rot on Manglietia decidua caused by Calonectria ilicicola in China. Plant Dis. 2022, 106, 1522. [Google Scholar] [CrossRef]
- Liu, G. List of Entry Phytosanitary Pests of the People’s Republic of China. Pestic. Mark. News 2007, 13, 38–39. [Google Scholar]
- Randall-Schadel, B.L.; Bailey, J.E.; Beute, M.K. Seed Transmission of Cylindrocladium parasiticum in Peanut. Plant Dis. 2001, 85, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Phipps, P.M. Control of Cylindrocladium black rot of peanut with soil fumigants having methyl isothiocyanate as the active ingredient. Plant Dis. 1990, 74, 438–441. [Google Scholar] [CrossRef]
- Lan, G.B.; He, Z.F.; She, X.M.; Tang, Y.F. Identification of the resistance to black rot disease in peanut (Arachis hypogaea) cultivars caused by Cylindrocladium parasiticum in Guangdong Province, China. J. Biosaf. 2016, 25, 214–217. [Google Scholar]
- Dewey, R.E.; Siedow, J.N.; Timothy, D.H.; Levings, C.S., 3rd. A 13-kilodalton maize mitochondrial protein in E. coli confers sensitivity to Bipolaris maydis toxin. Science 1988, 239, 293–295. [Google Scholar] [CrossRef]
- Corrêa, J.A.; Hoch, H.C. Identification of thigmoresponsive loci for cell differentiation in Uromyces germlings. Protoplasma 1995, 186, 34–40. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, H.F.; Zhang, Z.G.; Zheng, X.B. GTPase activating protein MoGcs1 is important for asexual development, appressorium differentiation and stress response in the rice blast fungus Magnaporthe oryzae. Acta Phytopathol Sin. 2016, 46, 17–26. [Google Scholar]
- Yakoby, N.; Beno-Moualem, D.; Keen, N.T.; Dinoor, A.; Pines, O.; Prusky, D. Colletotrichum gloeosporioides pelB\r, is an important virulence factor in avocado fruit-fungus interaction. Mol. Plant Microbe Interact. 2001, 14, 988–995. [Google Scholar] [CrossRef]
- Gao, F.; Chu, J.M.; Li, J.H.; Wang, M.L. Research progress in the pathogenesis of plant pathogenic fungi. Jiangsu J. Agric. Sci. 2014, 30, 1174–1179. [Google Scholar]
- Wang, Y.; Chen, J.; Li, D.W.; Zheng, L.; Huang, J. CglCUT1 gene required for cutinase activity and pathogenicity of Colletotrichum gloeosporioides causing anthracnose of Camellia oleifera. Eur. J. Plant Pathol. 2017, 147, 103–114. [Google Scholar] [CrossRef]
- Shang, Y.F.; Xiao, G.H.; Zheng, P.; Cen, K.; Zhan, S.; Wang, C.S. Divergent and Convergent Evolution of Fungal Pathogenicity. Genome Biol Evol. 2016, 8, 1374–1387. [Google Scholar] [CrossRef]
- Zhang, M.L.; Xiao, S.Q.; Zhang, L.; Zhang, Y.W.; Xue, C.S. Construction deletion vector of nonribosomal peptide Synthetases 6 of Setoshpaeria turcicum. J. Maize Sci. 2012, 20, 59–63. [Google Scholar]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Wit, P.J.G.M.D.; Mehrabi, R.; Burg, H.A.V.D.; Stergiopoulos, L. Fungal effector proteins: Past, present and future. Mol. Plant Pathol. 2009, 10, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. In planta transcriptome and proteome profiles of Spongospora subterranea in resistant and susceptible host environments illuminates regulatory principles underlying host–pathogen interaction. Biology 2021, 10, 840. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.D. Isolation and culture of fungi. In Plant Disease Research Methods, 2nd ed.; Fang, Z.D., Ed.; Agriculture Press: Beijing, China, 1979; pp. 112–136. [Google Scholar]
- Skovgaard, K.; Rosendahl, S.; O’Donnell, K.; Nirenberg, H.I. Fusarium commune is a new species identified by morphological and molecular phylogenetic data. Mycologia 2003, 95, 630–636. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.D.; Lee, S.B.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications, 1st ed.; Innis, M.A., Ed.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Silvestro, D.; Michalak, I. raxmlGUI: A graphical front-end for RaxML. Org. Divers. Evol. 2012, 12, 335–337. [Google Scholar] [CrossRef]
- Mawhorter, R.; Libeskind-Hades, R. Hierarchical clustering of maximum parsimony reconciliations. BMC Bioinform. 2019, 20, 612. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Yao, B.; Zhang, C. DFVF: Database of fungal virulence factors. Database 2012, 2012, bas32. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, A.; Belguith, H.; Ghram, I.; Hamida, J.B. LIPABASE: A database for ’true’ lipase family enzymes. Int. J. Bioinform. Res. Appl. 2011, 7, 390. [Google Scholar] [CrossRef]
- Lisitsa, A.V.; Gusev, S.A.; Karuzina, I.I.; Archakov, A.I.; Koymans, L. Cytochrome P450 Database. SAR QSAR Environ. Res. 2001, 12, 359–366. [Google Scholar] [CrossRef]
- Winnenburg, R.; Baldwin, T.K.; Urban, M.; Rawlings, C.; Köhler, J.; Hammond-Kosack, K.E. PHI-base: A new database for pathogen host interactions. Nucleic Acids Res. 2006, 34, 459–464. [Google Scholar] [CrossRef]
- Urban, M.; Pant, R.; Raghunath, A.; Irvine, A.G.; Pedro, H.; Hammond-Kosack, K.E. The Pathogen-Host Interactions database (PHI-base): Additions and future developments. Nucleic Acids Res. 2015, 43, 645–655. [Google Scholar] [CrossRef]
- Park, B.H.; Karpinets, T.V.; Syed, M.H.; Leuze, M.R.; Uberbacher, E.C. CAZymes Analysis Toolkit (CAT): Web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 2010, 20, 1574–1584. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Heijne, G.V.; Nielsen, H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Nielsen, H.; Brunak, S.; Heijne, G.V. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000, 300, 1005–1016. [Google Scholar] [CrossRef]
- Emanuelsson, O.; Brunak, S.; Heijne, G.V.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.X.; Meyer, C.A.; Wang, Q.; Liu, J.S.; Liu, X.S.; Zhang, Y. GFOLD: A generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 2012, 28, 2782–2788. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Luo, M.; Xiang, M.M.; Xiong, L.N.; Shu, Y.X.; Yu, G.H.; Dong, Z.Y.; Sun, Y.H. Comparative proteomic analysis of the moderately resistant and susceptible peanut cultivars during infestation by Cylindrocladium parasiticum. J. Plant Pathol. 2022. submitted. [Google Scholar]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar]
- Liu, H.H.; Wang, J.; Wu, P.H.; Lu, M.Y.; Li, J.Y.; Shen, Y.M.; Tzeng, M.N.; Kuo, C.H.; Lin, Y.H.; Chang, H.X. Whole-Genome Sequence Resource of Calonectria ilicicola, the Casual Pathogen of Soybean Red Crown Rot. Mol. Plant Microbe Interact. 2021, 34, 848–851. [Google Scholar] [CrossRef]
- Cao, C.J.; Xue, C.Y. More Than Just Cleaning: Ubiquitin-Mediated Proteolysis in Fungal Pathogenesis. Front Cell Infect. Microbiol. 2021, 11, 774613. [Google Scholar] [CrossRef]
- Liu, T.B.; Xue, C.Y. The Ubiquitin-Proteasome System and F-box Proteins in Pathogenic Fungi. Mycobiology 2011, 39, 243–248. [Google Scholar] [CrossRef]
- Han, Y.K.; Kim, M.D.; Lee, S.H.; Yun, S.H.; Lee, Y.W. A Novel F-Box Protein Involved in Sexual Development and Pathogenesis in Gibberella zeae. Mol. Microbiol. 2007, 63, 768–779. [Google Scholar] [CrossRef]
- Bartnicki-Garcia, S.; Bartnicki, D.D.; Gierz, G. Determinants of fungal cell-wall morphology: The vesicle supply center. Can. J. Bot. 1995, 73, 372–378. [Google Scholar] [CrossRef]
- Werner, S.; Sugui, J.A.; Steinberg, G.; Deising, H.B. A chitin synthase with a myosin-like motor domain is essential for hyphal growth, appressorium differentiation, and pathogenicity of the maize anthracnose fungus Colletotrichum graminicola. Mol. Plant Microbe Interact. 2007, 20, 1555–1567. [Google Scholar] [CrossRef]
- Zhang, J.J.; Jiang, H.; Du, Y.R.; Keyhani, N.O.; Xia, Y.X.; Jin, K. Members of chitin synthase family in Metarhizium acridum differentially affect fungal growth, stress tolerances, cell wall integrity and virulence. PLoS Pathog. 2019, 15, e1007964. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.R.; Koymans, L.; Kamataki, T.; Stegeman, J.J.; Feyereisen, R.; Waxman, D.J.; Waterman, M.R.; Gotoh, O.; Coon, M.J.; Estabrook, R.W.; et al. P450 superfamily: Update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 1996, 6, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Brink, H.M.V.D.; Gorcom, R.F.V.; Hondel, C.A.V.D.; Punt, P.J. Cytochrome P450 enzyme systems in fungi. Fungal Genet. Biol. 1998, 23, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Črešnar, B.; Petrič, Š. Cytochrome P450 enzymes in the fungal kingdom. Biochim. Biophys. Acta 2011, 1814, 29–35. [Google Scholar] [CrossRef]
- Shin, J.Y.; Bui, D.C.; Lee, Y.; Nam, H.J.; Jung, S.Y.; Fang, M.; Kim, J.; Lee, T.; Kim, H.; Choi, G.J.; et al. Functional characterization of cytochrome P450 monooxygenases in the cereal head blight fungus Fusarium graminearum. Environ. Microbiol. 2017, 19, 2053–2067. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.Y.; Chai, R.Y.; Qiu, H.P.; Jiang, H.; Mao, X.Q.; Wang, Y.L.; Liu, F.Q.; Sun, G.C. An S-(Hydroxymethyl) glutathione dehydrogenase is involved in conidiation and full virulence in the rice blast fungus Magnaporthe oryzae. PLoS ONE 2015, 10, e0120627. [Google Scholar] [CrossRef]
Figure 1.
Isolation and identification of the C. ilicicola Ci14017. (a) The CBR symptoms on peanut roots inoculated with Ci14017. (b) Phylogenetic tree established from the ITS region of Ci14017. The phylogenetic tree based on Maximum likelihood, Maximum parsimony and MrBayes analysis, the bootstrap support rate of the three is merged into the ML evolutionary tree. The arrow in the figure points to the branch where the bootstrap support rate is located.
Figure 1.
Isolation and identification of the C. ilicicola Ci14017. (a) The CBR symptoms on peanut roots inoculated with Ci14017. (b) Phylogenetic tree established from the ITS region of Ci14017. The phylogenetic tree based on Maximum likelihood, Maximum parsimony and MrBayes analysis, the bootstrap support rate of the three is merged into the ML evolutionary tree. The arrow in the figure points to the branch where the bootstrap support rate is located.

Figure 2.
Ci14017 gene functions categories. (a) GO classification statistics of Ci14017. Blast2GO (http://www.geneontology.org/), accessed on 28 April 2022 for GO term annotation; (b) KOG classification statistics of Ci14017. KOG (http://ww.ncbi.nlm.nih.gov/KOG/), accessed on 28 April 2022; and (c) gene statistics of KEGG pathway in Ci14017. KEGG pathway mapping (http://www.genome.jp/kegg/), accessed on 28 April 2022.
Figure 2.
Ci14017 gene functions categories. (a) GO classification statistics of Ci14017. Blast2GO (http://www.geneontology.org/), accessed on 28 April 2022 for GO term annotation; (b) KOG classification statistics of Ci14017. KOG (http://ww.ncbi.nlm.nih.gov/KOG/), accessed on 28 April 2022; and (c) gene statistics of KEGG pathway in Ci14017. KEGG pathway mapping (http://www.genome.jp/kegg/), accessed on 28 April 2022.


Figure 3.
Comparative genomics analysis of Ci14017. (a) Collinearity map of Ci14017 and Fusarium graminearum genomes. On the top is the F. graminearum genomes, and on the bottom is Ci14017, with secretory proteins marked in the red line. (b) Ci14017 hyperchronic evolution tree. The green numbers represent fossil ages. The blue numbers represent gene family expansion and contraction. The arrow in the figure points to the branch where the bootstrap support rate is located. (c) Venn analysis of gene families of 18 different genomes. The values of the black vertical bar chart indicated the number of homologous genes (including the specific genes of five genomes) in one to five genomes, while the values of the yellow horizontal bar chart indicate the total number of homologous genes in one genome with the other four genomes. The red dots represent the homologous genes of the five genomes. The blue dots represent the specific gene of Ci14017 and the green dots represent the homologous gene of C. canadiana cc13392.
Figure 3.
Comparative genomics analysis of Ci14017. (a) Collinearity map of Ci14017 and Fusarium graminearum genomes. On the top is the F. graminearum genomes, and on the bottom is Ci14017, with secretory proteins marked in the red line. (b) Ci14017 hyperchronic evolution tree. The green numbers represent fossil ages. The blue numbers represent gene family expansion and contraction. The arrow in the figure points to the branch where the bootstrap support rate is located. (c) Venn analysis of gene families of 18 different genomes. The values of the black vertical bar chart indicated the number of homologous genes (including the specific genes of five genomes) in one to five genomes, while the values of the yellow horizontal bar chart indicate the total number of homologous genes in one genome with the other four genomes. The red dots represent the homologous genes of the five genomes. The blue dots represent the specific gene of Ci14017 and the green dots represent the homologous gene of C. canadiana cc13392.


Figure 4.
Pathogenic gene analysis of Ci14017 effector protein. (a) PHI-base comparison results of 289 effector proteins. The text in the figure represents the phenotype after deletion of the mutant gene; (b) CAZymes classification of 1718 secreted proteins. CBMs: carbohydrate-binding modules, CEs: carbohydrate esterases, GHs: glucoside hydrolases, GTs: glycosyl transferases, PLs: polysaccharide lyases, AAs: auxiliary activities.
Figure 4.
Pathogenic gene analysis of Ci14017 effector protein. (a) PHI-base comparison results of 289 effector proteins. The text in the figure represents the phenotype after deletion of the mutant gene; (b) CAZymes classification of 1718 secreted proteins. CBMs: carbohydrate-binding modules, CEs: carbohydrate esterases, GHs: glucoside hydrolases, GTs: glycosyl transferases, PLs: polysaccharide lyases, AAs: auxiliary activities.

Figure 5.
Identification of 46 differentially expressed factor for optimal CBR development. (a) Mediate resistance traits of peanut T09 to Ci14017. (b) Venn analysis of 1146 genes matching the virulence-associated database. The values of the black vertical bar chart indicated the number of matching genes in one to seven databases, while the values of the yellow horizontal bar chart indicate the total number of matching genes in one database. The dots represent the genes that match the which database. (c) GO enrichment statistics of 46 proteins. Blast2GO (http://www.geneontology.org/), accessed on 5 May 2022 for GO term annotation. (d) KEGG pathway enrichment statistics of 46 proteins. KEGG pathway mapping (http://www.genome.jp/kegg/), accessed on 5 May 2022.
Figure 5.
Identification of 46 differentially expressed factor for optimal CBR development. (a) Mediate resistance traits of peanut T09 to Ci14017. (b) Venn analysis of 1146 genes matching the virulence-associated database. The values of the black vertical bar chart indicated the number of matching genes in one to seven databases, while the values of the yellow horizontal bar chart indicate the total number of matching genes in one database. The dots represent the genes that match the which database. (c) GO enrichment statistics of 46 proteins. Blast2GO (http://www.geneontology.org/), accessed on 5 May 2022 for GO term annotation. (d) KEGG pathway enrichment statistics of 46 proteins. KEGG pathway mapping (http://www.genome.jp/kegg/), accessed on 5 May 2022.

Figure 6.
Annotation and expression of 46 putative virulence-associated genes. ML tree of 46 putative virulence-associated genes. The different colors of the round spots indicate the matching results with each database. Transcriptome level gene expression calorimetry. P9ck: P562 9 days after water inoculation; PC9: P562 9 days after Ci14017 inoculation; T9ck: T09 9 days after water inoculation; TC9: T09 9 days after Ci14017 inoculation; PDA culture conditions. Proteome level gene expression calorimetry. PC_1, PC_2, PC_3: P562 9 days after Ci14017 inoculation; P9ck: P562 9 days after water inoculation; TC_1, TC_2, TC_3: T09 9 days after Ci14017 inoculation; T9ck: T09 9 days after water inoculation.
Figure 6.
Annotation and expression of 46 putative virulence-associated genes. ML tree of 46 putative virulence-associated genes. The different colors of the round spots indicate the matching results with each database. Transcriptome level gene expression calorimetry. P9ck: P562 9 days after water inoculation; PC9: P562 9 days after Ci14017 inoculation; T9ck: T09 9 days after water inoculation; TC9: T09 9 days after Ci14017 inoculation; PDA culture conditions. Proteome level gene expression calorimetry. PC_1, PC_2, PC_3: P562 9 days after Ci14017 inoculation; P9ck: P562 9 days after water inoculation; TC_1, TC_2, TC_3: T09 9 days after Ci14017 inoculation; T9ck: T09 9 days after water inoculation.

Figure 7.
Abundant conserved protein domain. Analysis of 46 virulence-associated genes domains. (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi?), accessed on 6 May 2022.
Figure 7.
Abundant conserved protein domain. Analysis of 46 virulence-associated genes domains. (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi?), accessed on 6 May 2022.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Basic information of Ci14017 whole genome.
| Ci14017 | Statistical Data |
|---|---|
| The number of scaffolds | 1993 |
| Assembly size (bp) | 66,310,315 |
| Mean length (bp) | 33,272 |
| Max length (bp) | 1,935,007 |
| Min length (bp) | 121 |
| Largest 10 total (bp) | 14,067,989 |
| The number of scaffolds (>1000 bp) | 820 |
| Scaffold N50 (bp) | 390,247 |
| Number of gaps | 442 |
| Total gap length (bp) | 244,347 |
| Mean gap length (bp) | 553 |
| Percent N’s | 0.37% |
| GC content | 48% |
| Repetitive DNA | 1.53% |
| tRNA | 423 |
| rRNA | 90 |
| Predicted coding genes | 18,366 |
| Total of exons | 53,680 |
| Coding region in genome | 42.23% |
| Mean gene length (bp) | 3610 |
| Mean coding gene length (bp) | 1525 |
| Mean exons per gene | 3 |
| Mean exon length (bp) | 522 |
Table 2.
Forty-six virulence-associated genes’ relevant information.
| SeqName | Description | Annotation to Categorize | Transcriptome/Fold Change (PC/TC) | Proteome/Fold Change (PC/TC) | Length |
|---|---|---|---|---|---|
| g11138.t1 | Cell division control protein 48 | Lipase/P450 | 0.645397155 | 3.817191977 | 836 |
| g11761.t1 | CVNH domain-containing protein | CAZymes | 1.077780219 | 2.576375314 | 409 |
| g12183.t1 | NADPH-cytochrome P450 reductase | Lipase/P450 | 0.777423319 | 2.274784162 | 690 |
| g12364.t1 | Peptidyl-prolyl cis-trans isomerase | / | 0.7462787 | 2.408002106 | 114 |
| g13131.t1 | alcohol dehydrogenase I | Lipase | 0.850951116 | 4.617233843 | 350 |
| g13442.t1 | Apoptosis-inducing factor 1 | P450 | 3.084508684 | 3.559369202 | 546 |
| g13926.t1 | Acetyl-coenzyme A synthetase | Lipase/P450 | 2.340024168 | 2.520666667 | 704 |
| g14087.t1 | S-(hydroxymethyl)glutathione dehydrogenase | Lipase | 0.35893699 | 3.679765396 | 888 |
| g14473.t1 | Fatty acid synthase subunit alpha | P450 | 1.920882975 | 2.612333333 | 1860 |
| g15360.t1 | 1,3-beta-glucanosyltransferase Gel1 | CAZymes | 1.406499686 | 2.461296413 | 453 |
| g15638.t1 | T-complex protein 1 subunit eta | Lipase | 0.933011105 | 2 | 557 |
| g15744.t1 | Glutamate decarboxylase | P450 | 0.89437205 | 3.28017126 | 571 |
| g15844.t1 | eIF4A-like protein | Lipase/P450 | 0.395142551 | 2.8717477 | 397 |
| g15879.t1 | CAMK/CAMK1/CAMK1-CMK protein kinase | Lipase/P450 | 1.036268466 | 2.70942813 | 370 |
| g15902.t1 | Carboxypeptidase Y A | Lipase/P450/Merops | 1.822742632 | 2.263530052 | 544 |
| g15950.t1 | Actin cytoskeleton-regulatory complex protein PAN1 | P450 | 0.418771014 | 1.666666667 | 1501 |
| g16158.t1 | Glucooligosaccharide oxidase | Lipase/P450/CAZymes/Secrete | 0.375885577 | 2.000333333 | 518 |
| g16583.t1 | endopolygalacturonase 1 | CAZymes/Secrete | 0.128246495 | 2.894074074 | 359 |
| g16828.t1 | probable endopolygalacturonase NFIA_008150 | CAZymes/Secrete | 0.758522499 | 1.909192825 | 382 |
| g17377.t1 | Thioredoxin reductase | Lipase | 0.46674335 | 2.260225252 | 321 |
| g18104.t1 | ubiquitin-conjugating enzyme E2-16 kDa | P450 | 0.804791205 | 1.666666667 | 146 |
| g18127.t1 | vacuolar protease A | P450/Merops | 1.864883004 | 2.301609848 | 502 |
| g18177.t1 | vesicle fusion factor | Lipase/P450 | 0.489926572 | 2.182333333 | 849 |
| g18328.t1 | elongation factor 3 | Lipase/P450 | 0.708137425 | 3.309420682 | 1055 |
| g18352.t1 | succinate-semialdehyde dehydrogenase (NADP+) | Lipase/P450 | 0.237659648 | 3.404154863 | 494 |
| g2659.t1 | chitin synthase | CAZymes | 1.141364798 | 1.666666667 | 1773 |
| g2660.t1 | chitin synthase | CAZymes | 1.075445163 | 1.999666667 | 1857 |
| g2688.t1 | Cytochrome c peroxidase, mitochondrial | CAZymes | 1.381410476 | 2.560020346 | 357 |
| g3217.t1 | catalase-peroxidase | CAZymes | 0.853030366 | 2.82071459 | 762 |
| g3305.t1 | NAD-binding Rossmann fold oxidoreductase family protein | CAZymes | 262.7 | 2 | 440 |
| g3805.t1 | GroES-like protein | Lipase/P450 | 0.450581519 | 2.705308465 | 357 |
| g4047.t1 | ubiquitin thiolesterase | Merops | 1.087713026 | 2 | 555 |
| g4370.t1 | GTP-binding protein rhoA | Lipase | 1.469419616 | 2.352234637 | 195 |
| g5045.t1 | Vanillin dehydrogenase | Lipase/P450 | 0.73931864 | 2.946907653 | 1021 |
| g5049.t1 | related to short chain dehydrogenase | Lipase/P450 | 0.908852984 | 2.114715806 | 258 |
| g5051.t1 | related to 6-hydroxy-D-nicotine oxidase | Lipase/P450/CAZymes | 0.727663175 | 2.707425743 | 461 |
| g5191.t1 | nitric oxide dioxygenase | P450 | 0.177916187 | 2 | 420 |
| g5581.t1 | Cytochrome P450 55A3 | Lipase/P450 | 0.595411287 | 1.999666667 | 404 |
| g5899.t1 | fungal specific transcription factor | Lipase/CAZymes/Secrete | 0.148888017 | 2.146453089 | 1191 |
| g6789.t1 | cysteine desulfurase | Lipase | 1.597153312 | 2 | 504 |
| g7130.t1 | glucosamine-fructose-6-phosphate aminotransferase | Merops | 0.508395005 | 3.672040022 | 702 |
| g7426.t1 | t-complex protein 1 subunit beta | Lipase | 0.053649734 | 2.256364713 | 540 |
| g7739.t1 | Peptidoglycan deacetylase | CAZymes | 43.97 | 1.666666667 | 335 |
| g7873.t1 | S-adenosyl-L-methionine-dependent methyltransferase | Lipase | 37.26 | 2 | 397 |
| g8661.t1 | predicted protein | P450 | 0.499370829 | 1.999666667 | 886 |
| g952.t1 | alcohol oxidase | P450/CAZymes | 0.340802231 | 3.471797575 | 688 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, X.; Luo, M.; Wu, W.; Dong, Z.; Zou, H. Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot. J. Fungi 2022, 8, 869. https://doi.org/10.3390/jof8080869
AMA Style
Chen X, Luo M, Wu W, Dong Z, Zou H. Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot. Journal of Fungi. 2022; 8(8):869. https://doi.org/10.3390/jof8080869
Chicago/Turabian StyleChen, Xinyu, Mei Luo, Wei Wu, Zhangyong Dong, and Huasong Zou. 2022. "Virulence-Associated Genes of Calonectria ilicola, Responsible for Cylindrocladium Black Rot" Journal of Fungi 8, no. 8: 869. https://doi.org/10.3390/jof8080869
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.