
Abstract
The childhood-onset or juvenile idiopathic inflammatory myopathies (JIIMs) are a heterogenous group of rare and serious autoimmune diseases of children and young people that predominantly affect the muscles and skin but can also involve other organs, including the lungs, gut, joints, heart and central nervous system. Different myositis-specific autoantibodies have been identified that are associated with different muscle biopsy features, as well as with different clinical characteristics, prognoses and treatment responses. Thus, myositis-specific autoantibodies can be used to subset JIIMs into sub-phenotypes; some of these sub-phenotypes parallel disease seen in adults, whereas others are distinct from adult-onset idiopathic inflammatory myopathies. Although treatments and management have much improved over the past decade, evidence is still lacking for many of the current treatments and few validated prognostic biomarkers are available with which to predict response to treatment, comorbidities (such as calcinosis) or outcome. Emerging data on the pathogenesis of the JIIMs are leading to proposals for new trials and tools for monitoring disease.
Key points
-
Juvenile idiopathic inflammatory myopathies (JIIMs) can differ from adult-onset myopathies in terms of the pathogenesis, autoantibody profile, disease phenotype and treatment response, but these differences need to be further defined.
-
The myositis-specific autoantibody and myositis-associated autoantibody profile of a patient can help to determine the disease phenotype and likely outcome of the patient, including their risk of having disease complications.
-
More research is needed to provide an evidence-based approach to the management of refractory JIIM, major organ involvement and myositis-related complications or comorbidities.
-
New therapeutic targets have been strongly implicated in JIIM by pathogenesis studies, most notably, the type I interferon pathway; clinical trials are urgently needed but innovative designs are required.
-
Further research is needed to identify specific dysregulated pathways in addition to type I interferon and how these pathways relate to the myositis-specific autoantibody or myositis-associated autoantibody clinical subtypes.
-
A better understanding is needed of the long-term outcomes of patients with JIIM into adulthood, including the factors that are important to patients and their families.
Similar content being viewed by others
Introduction
The childhood-onset or juvenile idiopathic inflammatory myopathies (JIIMs) are a group of rare but serious conditions of children and young people that predominantly affect the muscles and skin but can also involve other organs, including the lungs, gut, joints, heart and central nervous system. A newly defined European Alliance of Associations for Rheumatology–American College of Rheumatology (EULAR–ACR) system of classification1 captures the most prevalent group of JIIM, namely juvenile dermatomyositis (JDM). However, further refinement will be required for a classification that accurately captures the subtypes of JDM and delineates other forms of JIIM, including juvenile polymyositis, immune-mediated necrotizing myopathy (IMNM) in children or the overlap myositis syndromes. Unlike previous criteria, one advantage of the new EULAR–ACR criteria, according to an evaluation of these criteria in adult patients, is their ability to capture amyopathic dermatomyositis2. Although the EULAR–ACR classification criteria represent a new and superior standard, the Bohan and Peter criteria proposed in 1975 (ref. 3) have still been used in some recent literature.
An important advancement in the past 10 years is a greater understanding of the disease phenotype on the basis of the myositis-specific autoantibody (MSA) profile. MSAs, present in approximately 60% of children with JIIM4,5, can help to inform the disease course and risk of complications, such as interstitial lung disease (ILD) or calcinosis. MSA testing has helped to identify patients with IMNM, anti-synthetase syndrome or overlap syndromes who previously might have been classified as having juvenile polymyositis.
In terms of JIIM pathophysiology, vasculopathy and endothelial dysfunction are increasingly recognized as important elements, with number of circulating endothelial cells correlating with disease activity and nailfold abnormalities6. Type 1 interferon signature is a known key feature of JIIM but more work is needed to define the key drivers of this signature and the downstream effects that lead to immune dysregulation. Growing evidence supports involvement of mitochondrial dysfunction and endoplasmic reticulum (ER) stress. Greater understanding of pathogenesis might help to identify important therapeutic targets, shown most recently by the promise of JAK–STAT inhibition in the treatment of JIIM-related muscle, skin and lung disease7,8,9,10,11. This approach needs to be explored further by clinical trials.
In this Review, we describe the key features of JDM and its subtypes, as well as juvenile-onset IMNM, juvenile polymyositis and the overlap syndromes. We also discuss the clinical phenotypes of JIIM in relation to the MSA profile, highlighting the main clinical associations, response to treatment and caveats of antibody testing (Table 1). We review advances in our knowledge of the pathogenesis of JIIM and assess how evidence over the last decade has contributed to the understanding and management of these complex conditions, and what evidence is urgently needed to address the unmet needs in JIIM. Where data are available, we compare childhood-onset myositis and adult-onset myositis to highlight parallels or differences in antibody associations, genetics, clinical features, prognosis or outcomes. Detailed comparisons between adult and paediatric myositis have also been reviewed elsewhere12,13.
In the final section of this Review, we summarize current evidence in terms of JIIM treatment and highlight the need for head-to-head comparison studies to determine the best second-line treatment, options for recalcitrant disease and JIIM-related complications. A treatment algorithm for JIIM based on current available consensus is also presented, including the use of exercise therapy and psychological support, as well as medications. Finally, we discuss some of the key challenges in the management of JIIM and how international collaboration helps to overcome these challenges and improve our understanding of this rare but important group of diseases.
Epidemiology
JIIM has a reported incidence of between 1.6 and 4 cases per million children per year14 and an estimated prevalence of 2.5 cases per 100,000 children14, but limited data are available. Although the mortality in JIIM has decreased considerably since the pre-steroid era and is often reported as being below 4% worldwide15,16,17, mortality remains as high as 5–8% in some cohorts18,19,20. In a North American study, the mortality associated with juvenile connective tissue disease (CTD) overlap phenotypes was higher (standardized mortality ratio (SMR) 66.9) than that associated with juvenile polymyositis (SMR 30.7) or JDM (SMR 8.3)21. Risk factors, identified by multivariate analyses, included older age or illness severity at disease onset, weight loss and delay to diagnosis21.
As mortality rates have decreased over the years, more emphasis has been placed on evaluating long-term functional outcomes, morbidity and health-related quality of life. Notably, the risk of disease damage increases almost linearly for each year of disease22, highlighting the importance of early disease control. Damage, usually mild, is most common in the cutaneous, endocrine, muscular or skeletal domains, with identified predictors of damage, including high disease activity or severity of disease at onset, duration of active disease, the presence of early organ damage (within 6 months of diagnosis) and functional disability19,23,24. Functional impairment is usually mild, but reported in up to 41% of patients and can be associated with increased pain and decreased quality of life16,17,19,20,23,24,25. Children can be affected by impaired growth or delayed puberty, particularly if there is preceding growth failure or if the active phase of disease occurs during early puberty26. In a report of adults who had JDM and were surveyed at an average age of 20 years, 59% perceived that their myositis was still active and 65% were still taking immunosuppressive medication27. JIIM is also associated with long-term risks relating to cardiovascular, pulmonary or cerebrovascular disease6,28,29,30,31.
Clinical phenotypes
On the basis of clinical and histopathological findings, JIIM can be separated into various subtypes. JDM, the most common JIIM subtype, represents more than 80% of patients, followed by overlap myositis14,15,19,32. In this section, we first review the clinico-serological subtypes of JDM before discussing the features of amyopathic JDM, anti-synthetase syndrome, IMNM and overlap syndromes. In the absence of myositis, patients with characteristic skin rashes are considered to have amyopathic or clinically amyopathic JDM, but this phenotype is rare in children33,34,35. Juvenile polymyositis is a very rare subtype, characterized by severe muscle inflammation and characteristic but not pathognomonic histological, radiological and electromyographic findings36. Emerging data suggest that some patients previously diagnosed as having JDM or juvenile polymyositis instead fall within the IMNM37, overlap myositis or anti-synthetase syndrome category38, on the basis of their autoantibody profile (Table 1).
Juvenile dermatomyositis
JDM is defined by the presence of proximal symmetric myositis and characteristic cutaneous features and has a median age at diagnosis of 7.4 years32. Calcinosis has been reported in 20–47% of patients with JDM in different cohorts16,39. Approximately 60% of patients with JDM are positive for a myositis-specific antibody (MSA) (Table 1). Increasingly, expert consensus is that JDM can be divided into the following subtypes defined by the presence of a specific MSA: anti-Mi2 antibody-positive JDM, anti-nuclear matrix protein 2 (NXP2) antibody-positive JDM, anti-transcriptional intermediary factor 1 (TIF1) antibody-positive JDM, anti-melanoma differentiation-associated gene 5 (MDA5) antibody-positive JDM, anti-small ubiquitin-like modifier activating enzyme (SAE) antibody-positive JDM, and MSA-negative JDM38 (Table 1). Two MSAs can coexist in the same patient, but this is extremely rare, although some patients do have both an MSA and one or more myositis-associated autoantibodies (MAAs)5.
Anti-TIF1 antibody-positive JDM
Anti-TIF1 antibodies are the most common MSA in JIIM, with a reported frequency of between 17% and 35% (refs. 4,5,40) (Table 1). These antibodies are most common in white children and those children with a younger age at disease onset5 (median age of 7 years at disease onset in one North American study). The clinical phenotype of anti-TIF1 antibody-positive JDM includes mild muscle disease with relatively low creatinine kinase serum levels but with severe skin involvement, including an increased risk of ulceration and lipodystrophy4,5,41. Other frequent skin manifestations include Gottron papules, malar rash, erythema, ‘shawl-sign’ rash, photosensitivity and cuticular overgrowth5. Dysphagia can also occur in these patients4. Some patients have a chronic severe disease course, requiring the use of second-line or third-line treatment regimes, including cyclophosphamide or biologic drugs4. Although anti-TIF1 antibodies confer an increased risk of malignancy in patients with adult-onset myositis42,43, this association has not been reported in individuals with childhood-onset myositis.
Anti-NXP2 antibody-positive JDM
Anti-NXP2 antibodies (initially known as anti-MJ antibodies) are present in approximately 15–25% of patients with JDM (Table 1) and are one of the most common MSAs in white populations5,44,45. Anti-NXP2 antibody-positive patients with JDM typically present at a young age, and have the highest incidence of calcinosis among the various JDM antibody subtypes, with age at onset itself found to be linearly associated with risk of calcinosis in a UK cohort46. Calcinosis is also associated with the presence of anti-NXP2 antibodies in adult-onset idiopathic inflammatory myopathy (IIM)47. Muscle disease can be severe in childhood-onset anti-NXP2 antibody-positive disease and can include muscle contractures, muscle atrophy and functional compromise45. Other features of anti-NXP2 antibody-positive disease include gastrointestinal involvement, risk of dysphagia, dysphonia and skin ulceration4. The disease can be difficult to treat, has a low probability of treatment discontinuation, does not always respond well to conventional treatment and can result in a poor long-term prognosis48,49.
Anti-MDA5 antibody-positive JDM
Patients with anti-MDA5 antibody-positive JDM typically have minimal or no muscle involvement4,50,51. The characteristic clinical phenotype includes frequent skin rashes, cutaneous ulceration and arthritis (affecting mainly the small joints of the hand and feet), in addition to constitutional symptoms (such as weight loss), oral ulceration and increased risk of ILD4,50,52,53,54. Patients with early ILD detected by computerized tomography or pulmonary function tests are frequently asymptomatic55. Rapidly progressive ILD is a rare but potentially fatal complication of IIM in both children and adults with anti-MDA5 antibodies and reports suggest that this complication occurs more often in East Asian populations than in other populations55,56,57. Anti-MDA5 antibody-positive patients are more likely to receive short-term treatment with glucocorticoids than patients with other JIIM autoantibody subtypes, although overall treatment duration and frequency of clinical remission in anti-MDA5 antibody-positive JDM is similar to that of other JDM subtypes50.
Anti-Mi2 antibody-positive JDM
Anti-Mi2 antibodies are present in 4–10% of patients with JDM4,5. Anti-Mi2 antibody-positive JDM is more common in Hispanic patients with an older disease onset (median age of disease onset of 11 years) than other JIIM autoantibody subtypes5. Children with anti-Mi2 antibody-positive JDM typically present with severe muscle disease and notable skin involvement, frequently referred to as ‘classic JDM’4,5. Common skin rashes include those pathognomonic for JDM (such as heliotrope rash and Gottron papules), along with malar rash and periungual nailfold capillary abnormalities5. The severity of the myositis is reflected by the high muscle biopsy scores of the patients58. Children with anti-Mi2 antibody-positive JDM are less likely to have ILD than patients with other JDM subtypes, but are at a greater risk of dysphagia and oedema4,5. Despite the severe presentation, anti-Mi2 antibody-positive patients respond well to conventional treatment and have a good chance of being off treatment at 2 years48.
Amyopathic juvenile dermatomyositis
Amyopathic JDM can occur in some children but it is rare (<5% of patients with JIIM)35,59. Anti-TIF1 antibodies, followed by anti-MDA5 antibodies, are the most common MSAs associated with this JIIM subtype34. Patients with amyopathic JDM tend to have a young age of disease onset and have less myalgia, arthritis, calcinosis, dysphagia or abdominal pain than other patients with JDM34. Skin manifestations include Gottron papules, heliotrope rash, malar rash, periungual capillary abnormalities and photosensitivity34. Some patients with anti-SAE antibodies can present initially with skin disease, with muscle involvement occurring at a later stage4,60. In a case report, one patient had anti-SAE antibody-positive amyopathic JDM complicated by ILD61. In the absence of myositis, some experts believe that the presence of an MSA can support a diagnosis of JIIM62,63.
Anti-synthetase syndrome
Anti-synthetase syndrome is characterized by the presence of antibodies against aminoacyl tRNA synthetases (anti-ARS antibodies; also known as anti-synthetase antibodies) and a broad spectrum of clinical features. Eight anti-synthetase antibodies have so far been described in IIM: anti-Jo1 (anti-histidyl-tRNA synthetase), anti-PL12 (anti-alanyl-tRNA synthetase), anti-PL7 (anti-threonyl-tRNA synthetase), anti-EJ (anti-glycyl tRNA synthetase), anti-KS (anti-asparaginyl-tRNA synthetase), anti-OJ (anti-isoleucyl-tRNA synthetase), anti-Ha (anti-tyrosyl-tRNA synthetase) and anti-Zo (anti-phenylalanyl-tRNA synthetase) antibodies. Clinical manifestations of anti-synthetase syndrome, as documented in a North American study, include proximal muscle weakness (100%), arthritis (74%), mechanic’s hand (32%), fever (63%), Raynaud phenomenon (32%) and ILD (63%)5. Anti-synthetase syndrome is rare in children and much knowledge is extrapolated from the disease in adults. Among adults with anti-synthetase syndrome, patients positive for anti-Jo1 antibodies are more likely to have myositis, whereas other patients, especially those with anti-PL12 antibodies, are more likely to have isolated ILD and therefore might present initially to a respiratory physician64,65.
Immune mediated necrotizing myopathy
IMNM is a rare and recently characterized subtype of JIIM that includes anti-signal recognition particle (SRP) antibody-positive myopathy, anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) antibody-positive myopathy and antibody-negative IMNM38,66. The hallmark muscle biopsy finding of IMNM is muscle fibre necrosis with the absence or minimal presence of lymphocytic infiltrate66. Children with IMNM characteristically present with severe muscle weakness and notably elevated serum levels of muscle enzymes. Anti-SRP antibody-positive patients can have dysphagia67, and in rare instances can have cardiac involvement66,68. Some patients can also present with skin and other extra-muscular manifestations, which can include arthralgia or Raynaud phenomenon, as well as ILD66. Children with anti-HMGCR antibodies typically present with severe proximal muscle weakness, and can have muscle atrophy, contractures and arthralgia37,66. Although in adults the development of anti-HMGCR antibodies is frequently associated with exposure to statins, this association is absent in children with anti-HMGCR antibodies37,69. Autoantibody negative IMNM remains poorly characterized.
In children, anti-SRP or anti-HMGCR myopathy presenting with slowly progressive muscle weakness could be mistaken for muscular dystrophy70. If there is a high index of suspicion with muscle biopsy, immunohistochemical or genetic testing might be appropriate. Patients with muscular dystrophy can share the same pattern of muscle weakness (with more proximal than distal involvement), elevation of muscle enzymes, oedema on MRI and myopathic features on biopsy, but can be distinguished from JIIM by a tendency to have a more insidious disease onset, weakness in other muscle groups, calf muscle of generalized muscle hypertrophy, joint contractures, scapular winging, scoliosis, spinal rigidity, cardiomyopathy or macroglossia and the absence of an MSA70.
Overlap myositis
Currently, no unifying internationally accepted definition of overlap myositis exists as different CTDs can have similar clinical features. An international survey of clinical opinion on criteria for JDM–scleroderma overlap, which occurs in 15–20% of patients with JDM according to some reports71, proposed the use of the presence of two or more of the following criteria: Raynaud phenomenon, sclerodactyly and sclerodermatous skin changes in a child fulfilling criteria for JDM72. In a large US study of 1,718 patients with systemic lupus erythematosus (SLE) (451 paediatric and 1,267 adult patients), 6.3% of the patients had concurrent myositis73, whereas in a UK cohort of patients with JIIM, 2.5% of the patients were given a diagnosis of JDM–SLE overlap15.
The most commonly detected autoantibodies in overlap syndromes are MAAs (Table 1), although these antibodies can also be found in other JIIM subtypes. One or more MAAs might co-occur with MSAs in the same patient4,12. MAAs include anti-Ro52, anti-PM/Scl and anti-U1RNP antibodies5,74. For example, in one cohort, MSAs were detected in 6/49 (12%) of patients with overlap CTD or mixed CTD, whereas MAAs were present in 25/49 (51%) of the patients4. Overlap syndromes are associated with an increased risk of extra-muscular manifestations and a higher risk of mortality, in particular because of the higher risk of ILD, compared with other JIIM autoantibody subtypes, highlighting the importance of a correct diagnosis and early treatment32.
Myositis in other paediatric conditions
Other than primary myositis, myopathy or myositis can be a presenting feature in a number of different inflammatory conditions seen in childhood. Clinical presentation of myositis in childhood sarcoidosis is a rare but reported manifestation75. Thus, sarcoidosis or granulomatous myositis should be considered in patients presenting with myositis and hypercalcaemia75. Myositis can also be present in childhood vasculitides, with reports of polyarteritis nodosa presenting as polymyositis76 and deficiency of adenosine deaminase 2 (DADA2), a monogenic autoinflammatory disease, presenting with inflammatory myositis77.
Advances in genetic testing have resulted in an increasing recognition of monogenic autoinflammatory diseases and testing for such diseases should be included in the differential diagnosis of patients with myositis78. Characteristic features of monogenic autoinflammatory diseases include onset at an early age, fever and systemic inflammation affecting the eyes, joints, skin and serosa, but any system can be involved. Monogenic interferonopathies, such as proteasome-associated autoinflammatory syndromes and stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy syndrome, can mimic JDM78,79. Protracted febrile myalgia is a rare manifestation of familial Mediterranean fever characterized by prolonged severe and symmetric muscle pain, fever and elevated inflammatory markers that can also mimic JIIM80.
Pathophysiology
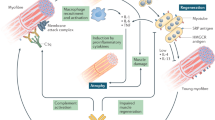
Although the triggers of disease in JIIM remain elusive, several studies over the past few years have implicated new or interconnected mechanisms in the skin, blood vessels and muscle (Fig. 1), as discussed in this section.

The pathogenesis of juvenile idiopathic inflammatory myopathy (JIIM) involves a complex interplay between genetic and environmental factors, leading to immunological, vascular and metabolic dysfunction. a, Environmental triggers of JIIM might include ultraviolet (UV) radiation, pollution and microbial infections. b, Genetic loci in the MHC and non-MHC regions are implicated in disease susceptibility and development. c, Type I interferon signalling is thought to have a central role in the pathological changes seen in various tissues. d, Immune dysregulation within the skin, muscle and blood vessels, as well as in other tissues (not shown), is thought to contribute to disease. Within the muscle, the overexpression of MHC proteins, a hallmark feature thought to be driven by interferons, contributes to endoplasmic reticulum (ER) stress leading to an inflammatory cascade via the nuclear factor kappa B (NF-κB) pathway. Autoreactive B cells are present, as demonstrated by the production of myositis-specific antibodies (MSAs), and regulatory B (Breg) cells have a pro-inflammatory phenotype (including producing elevated levels of IL-6). Circulating inflammatory mediators include Galectin-9 and CXCL10, which correlate with disease activity. Abnormalities in the small blood vessels are reflected by a high number of circulating endothelial cells, which correlates with disease activity; muscle capillary loss and complement deposition on capillaries also frequently occur. T cell dysfunction includes a skewing of the T cell compartment towards a T helper 17 (TH17) cell phenotype, including within the follicular helper T (TFH) cell population. Both neutrophil extracellular trap (NET) formation and mitochondria dysfunction occur in JDM and might be part of a pathological loop that drives interferon production. Overall, the pathogenesis of JIIM involves a complex interplay between innate and adaptive immunity that affects muscle, skin and vascular tissues to drive ongoing inflammation and tissue damage. ISGs, interferon-stimulated genes.
Environmental risk factors
Although the exact cause of this heterogeneous group of diseases remain largely unknown, complex interactions between genetic and environmental factors, as well as immune and non-immune mechanisms, have a role in JIIM pathogenesis81. The contributions of several bacterial and viral pathogens have been studied, including streptococcal infections, picornavirus, enterovirus, mycoplasma, with inconclusive results82,83,84,85. Some patients have presented with myositis following SARS-CoV-2 infection or vaccination against SARS-CoV-2 (refs. 86,87), but such case reports await confirmation by larger epidemiological studies. Ultraviolet light intensity and exposure have been associated with both disease aetiology and severity in JIIM88,89,90. Such exposures might be associated with specific clinico-serological subtypes: for example, in a large North American study, previous high exposure to ultraviolet light was associated with increased odds of having anti-TIF1 antibodies89. Other studied risk factors in JIIM include air pollution, maternal smoking and maternal occupation91. Some evidence suggests that certain immunizations, stressor events, heavy exercise prior to diagnosis and prolonged breastfeeding increase the risk of specific JIIM phenotypes92 but the results need to be confirmed in larger multinational studies.
Genetics
In both adult and paediatric IIM, the strongest genetic association in white populations is within the 8.1 ancestral haplotype (AH8.1; also known as the HLA A1-B8-DR3-DQ2 haplotype) of the major histocompatibility complex (MHC), first detected by GWAS analyses93. Subsequent studies using Immunochip data in well-characterized, larger cohorts have confirmed this association94. More recently, in 2022, in a large international genetics study of JDM that used dense exome SNP genotyping, researchers revealed that the allele HLA-DRB1*03:01 and amino acid position 37 within HLA-DRB1 are both strongly associated with JDM, and that this association was independent of position 74, a position associated with adult-onset dermatomyositis, enabling differentiation between juvenile and adult-onset disease95. Further analyses suggested that position 37 of HLA-DRB1 was independent of the AH8.1 ancestral haplotype and confirmed previous associations with AH8.1 and HLA-DRB1*03:01. Specific associations of the HLA-DQB1*02 allele with disease differ between adult-onset and childhood-onset anti-TIF1 antibody-positive dermatomyostis96. Similarly, paediatric-onset anti-HMGCR antibody-positive myositis has a specific association with HLA-DRB1*07:01, whereas adult-onset anti-HMGCR antibody-positive myositis is associated with HLA-DRB1*11:01 (ref. 37). Other non-MHC genetic loci, including the R620W variant of PTPN22 and a non-synonymous SNP (rs2304256) in TYK2, have also been associated with both adult and juvenile IIM, as well as other autoimmune conditions97,98.
Vasculopathy of JIIM
Vasculopathy and endothelial dysfunction are thought to have an important role in JDM and have been associated with systemic disease99. In a flow cytometry analysis, the number of circulating endothelial cells but not circulating endothelial progenitor cells were increased in the peripheral blood of patients with JDM compared with healthy individuals100; a study of 90 patients with JDM found that the number of circulating endothelial cells correlated with disease activity and nailfold abnormalities, and were increased in both patients with active JDM and patients with inactive JDM compared with healthy individuals6. In a separate study, patients with JDM who were positive for anti-TIF1 antibodies had lower nail fold end row loop counts (indicative of vasculopathy) at diagnosis and a prolonged duration of untreated disease, compared with other patients with JDM101. Endothelial soluble adhesion molecules, including soluble intercellular adhesion molecule 1 (sICAM1) and sICAM3, soluble vascular cell adhesion molecule 1 (sVCAM1), VCAM1 and E-selectin, are key players in the adhesion and migration of leukocytes through the endothelium towards inflamed sites and are under investigation as biomarkers of vasculopathy in JIIM6,99,102. These molecules are mainly secreted by activated endothelial cells, highlighting the association of these molecules with vasculopathy in JDM. The soluble forms of these molecules maintain many of the functions and the structure of the cell-bound adhesion molecules and are therefore of interest as potential therapeutic targets.
The role of interferon and immune cells
A strong interferon type I signature has been extensively implicated as a characteristic feature of JIIM, including studies of patient blood, muscle and skin79,103,104,105. Type II interferon has also been associated with JIIM106. Both type I and II interferons originate as viral interfering proteins; several type I interferons exist (including IFNα and IFNβ), all of which bind to the type I interferon receptor107, whereas IFNγ is the only type II interferon and binds to the separate type II interferon receptor. Several different assays exist that assess the levels of interferon types I and II, the downstream targets and related biomarkers (Table 2).
In parallel to the interferon pathway, both innate and adaptive immune dysregulation are thought to contribute to JIIM. The presence of MSAs and their association with distinct clinical phenotypes (which differ between juvenile and adult-onset disease52) strongly implicate a role for B cells in disease. Notably, in an international trial of adult and juvenile IIM, B cell depletion appeared to have clinical benefit in patients with JDM, according to a sub-analysis108. In additional to clinical phenotypes, specific MSAs are also associated with pathological conditions and patterns of inflammatory infiltrate in muscle biopsy samples58. In a study of CXCR5+ follicular helper T (TFH) cells in patients with JDM, the cells were skewed towards T helper 2 (TH2) and T helper 17 (TH17) cell phenotypes109, which might drive B cells towards autoantibody production and a pro-inflammatory phenotype. A separate study confirmed skewing of the T cell compartment towards a TH17 phenotype in juvenile, adolescent and adult patients with dermatomyositis110. Inflammatory T cells, B cells and tissue macrophages are all present in the inflamed muscle of patients with JDM111,112,113. An analysis of peripheral blood B cells in patients with JDM showed that a population of immature transitional B cells (CD19+CD24hiCD38hi cells) is expanded during active disease and correlates with disease activity114. Transcriptional and functional analyses have confirmed that these immature transitional B cells have an upregulated IFNα signature that is associated with an abnormal ratio of IL-6 to IL-10 production, suggesting that these cells are driven towards a pro-inflammatory phenotype that hinders the immunoregulatory properties of the cells114.
The inflammatory T cell and B cell infiltrate within muscle biopsy samples (which is typically perivascular) correlates with interferon-driven MxA expression and drives the inflammatory domain score of a JDM muscle biopsy score48,103,111. This muscle biopsy score has prognostic value in predicting treatment and disease48,111. Tissue macrophages in JDM muscle are highly pro-inflammatory and secrete both cytokines and pro-inflammatory molecules, including calprotectin112. In an immunofluorescence analysis of muscle biopsy samples, the expression of IFNγ and HLA class II molecules was increased in patients with JDM not undergoing treatment compared with healthy individuals, and the type I and type II interferon scores were associated with muscle infiltration by endomysial and perimysial CD3+ cells, as well as with CD68+ cells, and perifascicular atrophy of the muscle106. Transcriptomic analyses suggest that skin lesions of patients with JDM contain higher numbers of macrophages and CD4+ memory T cells than non-lesional skin and share a similar gene expression pattern to skin lesions from patients with childhood-onset SLE115, including a prominent type I interferon signature. However, the factors most important in driving the type I interferon signature and immune cell dysregulation remain elusive. More work is needed to understand these mechanisms: high-resolution techniques (such as single-cell transcriptional analyses by RNA sequencing) for assessing skin, muscle and blood samples, as well as differential transcriptional expression in specific cell lineages, in parallel with functional studies in JDM, are ongoing and will generate important mechanistic insights into the interferon signature, its relation to other dysregulated pathways and how these processes are impacted by treatment or disease activity.
Neutrophils, NETs and mitochondrial dysfunction
Neutrophils, an essential component of the innate immune system, can produce neutrophil extracellular traps (NETs) that comprise DNA–histone complexes and other released proteins. The role of NETs is to help to capture, degrade and kill pathogens (such as bacteria)116. Various studies implicate dysregulated neutrophil pathways, including NET formation, in JDM. For example, a muscle biopsy analysis found increased amounts of NET remnants in patients with JDM compared with healthy individuals, which was more evident in patients with calcinosis117. In a concurrent study, the level of circulating NET complexes was also higher in patients with JDM than in healthy individuals118 and correlated with disease activity and the presence of anti-MDA5 antibodies, but conversely did not correlate with calcinosis117,118. In one of these studies, NETs were shown to contain mitochondrial DNA (mtDNA)117, which is notable as studies in SLE have shown that mitochondrial dysfunction leads to the extrusion of oxidized mtDNA in NETs, which in turn induces an type I interferon response119. Indeed, gene expression network analysis of muscle has implicated a role for mitochondrial dysfunction in JDM, and a recent study demonstrated that abnormal mitochondrial function in monocytes (including the presence of enlarged mitochondrial networks or ‘megamitochondria’) in patients with JDM leads to the production of oxidized mitochondrial DNA and drives further type I interferon production120,121. Furthermore, anti-mitochondrial autoantibodies are present in the serum of some patients (1% (4/371) of patients in one enzyme-linked immunosorbent assay (ELISA)-based analysis)122. This growing body of evidence supports the involvement of mitochondrial dysfunction in JDM pathogenesis and in type I interferon-mediated inflammation.
ER stress
JDM is characterized by an increased expression of MHC class I molecules on muscle fibres, which is thought to be driven by both type I and type II interferon signalling103,123. Accumulation of class I MHC proteins can result in ER stress and can lead to cell death124. ER stress might also synergise with factors secreted by infiltrating myeloid cells, such as myeloid-related protein 8 (MRP8), MRP14 and other endogenous TLR ligands, to further damage the muscle112. For example, in one study, concentrations of MRP8–MRP14 complexes were significantly increased in the serum of patients with JDM compared with age-matched healthy controls (P > 0.05); further analysis suggested that these inflammatory proteins were secreted by CD68+ myeloid cells and synergized with ER stress to promote the production of IL-6 and MCP1 in the muscle112. In a separate muscle biopsy analysis, the muscles of adults with IIM contained higher levels of proteins involved in the ER stress-induced-autophagy pathway (such as the ER chaperone protein glucose-regulated protein 78 (GRP78)) than muscles of individuals lacking any myopathic features, which correlated with levels of autophagy, muscle damage and disease activity125. These studies demonstrate that ER stress might have an important role in JIIM pathogenesis.
Diagnosis
The diagnosis of JIIM requires careful evaluation of a number of clinical features, supported by a combination of laboratory, radiological and histopathological investigations. A Single Hub and access point for Paediatric Rheumatology in Europe (SHARE) initiative-based consensus guideline has set out recommendations for the diagnosis of JIIM, including investigations to differentiate JIIM from other causes of muscle weakness, to confirm a diagnosis of JIIM and to determine the presence of organ involvement126. A similar process has been followed by the Paediatric Rheumatology Association of Japan and the Japan College of Rheumatology to produce a clinical practice guideline, recognizing that the frequency of complications and drug use differs among Europe, the USA and Japan127. Diagnostic testing has been discussed in detail elsewhere85, and therefore a full description of diagnostic work-up will not be repeated here but might include formal evaluation of muscle strength, detailed cutaneous assessment, testing for muscle enzymes or other parameters in the blood and performing pulmonary function tests, electrocardiography, echocardiography and radiological investigations. In this section we discuss notable changes in practice over the past decade, particularly relating to the role of MRI, muscle biopsy and MSAs85,126.
MRI is now favoured as a diagnostic tool, but muscle biopsy also remains important, particularly in the absence of skin rash or when presentation is atypical126. When performed, use of a standardized JDM biopsy score is helpful in quantifying the severity of histopathological abnormalities, and together with MSA status, might help to predict the disease course48,111,126.
A major advance over the last decade has been the routine use of MSAs to aid the diagnosis of JIIM, to help to define or predict disease phenotype and to develop a more personalized approach to management64,128,129 (although the absence of an MSA does not rule out JIIM5,128,130). A recent survey of members of the International Myositis Assessment and Clinical Studies (IMACS) group found that over 80% of participants reported that MSA testing increased their confidence in the diagnosis and information that they gave to patients on prognosis128. However, more than 90% of respondents expressed the need for more education on the interpretation of antibody results128. The MSA–MAA profile might influence the investigative screening or treatment decisions by indicating the risk of a chronic disease course or specific complications such as ILD or calcinosis (Table 1). Results of MSA–MAA testing can vary depending on which technique is used, with some techniques not reliably detecting certain MSAs, as described in Table 1. Measurement by immunoprecipitation is considered the gold standard, but is expensive and time consuming and additional testing is required to differentiate between the presence of anti-NXP2 antibodies and anti-MDA5 antibodies131. Other techniques used in practice include line blot, dot blot, commercial multiplex assays, ELISAs and gel precipitation. Line blot is a technique that is cheap and rapid to perform, but false positives can occur, and this technique does not reliably detect anti-TIF1 (ref. 130) antibodies, which is the most common MSA in JIIM130,131,132. ELISA is a reliable test for detecting anti-TIF1 antibodies and produces a fast and quantitative result, but multiple assays might be required to test for all MSAs. Some MSAs are cytoplasmic and therefore MSAs can still be present when an antinuclear antibody (ANA) test result is negative. The staining pattern seen on human epithelial type 2 (HEp-2) cells can be used, along with the clinical phenotype, to help ascertain if the results of MSA testing are correct131. False-positive results should be considered if more than one MSA is reported as positive, or if the MSA result does not fit with the HEp-2 staining pattern or expected clinical phenotype. Repeating a test using the same technique is rarely useful and in ambiguous cases a different testing technique or specialist laboratory is preferable. Further details on the expected HEp-2 cells staining pattern and challenges with MSA testing are summarized in Table 1.
Management
The treatment of JIIM needs to consider the disease severity of the patient, including the presence of systemic and/or organ involvement and the disease phenotype. As well as these features, the MSA–MAA profile can inform the management and treatment of the patient, given the associations of specific MSAs and MAAs with clinical phenotypes, prognosis and risks of complications (Fig. 2). Treatment decisions are best made in a specialist paediatric centre by a multidisciplinary team, owing to the rarity and heterogenicity of the diseases126,133. Consensus guidelines provide a framework for health care professionals on the basis of the best possible evidence available126,133. A 2022 evidence-based British Society for Rheumatology guideline for childhood and adult-onset myositis, and a previous European consensus recommendation for JIIM, emphasize the need for a safe and effective exercise programme and attention to psychological wellbeing in addition to drug therapies for the management of JIIM126,133. The Childhood Arthritis and Rheumatology research Alliance (CARRA) guideline provides Consensus Treatment Plans for different severity levels of juvenile myositis134,135,136. Treatments for JIIM have been well described in reviews elsewhere85,129,137,138. A suggested treatment algorithm based on the best available current evidence and integrating current recommendations from the various guidelines is shown in Fig. 3. In this section, we outline various drug-related and non-medication-related aspects in the management of JIIM.

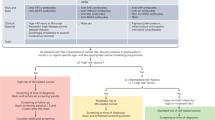
Owing to the rarity and heterogeneity of juvenile idiopathy inflammatory myopathy (JIIM), children and young people should be managed by a multidisciplinary team in a specialist centre. To predict the severity of the disease and the potential need for treatment escalation, many factors are considered, as illustrated, including the presence or absence of severe muscle weakness, dysphagia, ulcerative skin disease or major organ involvement. The myositis-specific autoantibody (MSA) and/or myositis-associated autoantibody (MAA) profile might predict the risk of JIIM-related complications, including major organ involvement. Some features associated with specific MSAs or MAAs are shown, but specific complications are not exclusive to patients with these MSA–MAA profiles and not all patients with a particular MSA–MAA profile will demonstrate these complications. CMAS, childhood myositis assessment scale; GI, gastrointestinal; ILD, interstitial lung disease; IMNM, immune-mediated necrotizing myopathy; MMT-8, manual muscle testing in eight muscle groups.

A treatment algorithm for juvenile idiopathic inflammatory myopathy (JIIM) is presented, based on evidence-informed consensus recommendations in UK and Europe126,133. Treatments need to be individualized and include consideration of the patient age, preferences for oral or parenteral administration of medications, severity of disease and response to treatment. No single approach will be right for every patient and clinicians need to use best judgement on the basis of evidence available. In most cases, with the exception of randomized controlled trials evaluating methotrexate versus ciclosporin, rituximab or exercise in myositis, evidence is limited to case series or cohort studies. More research is needed to compare the efficacy of second-line or third-line treatment options and determine the best treatment approach for myositis-related complications such as interstitial lung disease (ILD) or calcinosis. More evidence is also needed to determine the best treatment for refractory disease, which can be defined as myositis that responds inadequately to at least two immunosuppressant or immunomodulatory drugs given in their full dose for a minimum of 3 months, hindering weaning of corticosteroid. Patients with JIIM should have regular reviews that include measurement of muscle strength, assessment of skin disease and extra-muscular manifestations. Adherence to medication should be checked if patients fail to respond as expected. Treatment should be escalated if patients fail to respond adequately to treatment or are intolerant to the treatment. Exercise therapy and psychological support are important aspects to the management of JIIM in addition to medication. IVIG, intravenous immunoglobulin; JAK, janus kinase; MMF, mycophenolate mofetil.
Medication
A combination of high-dose corticosteroid in combination with methotrexate (15–20 mg/m2, maximum 40 mg/week) is the first-line induction treatment for most cases of JIIM126,133,139. Methotrexate is favoured over ciclosporin as it has a more favourable adverse effects profile; however, both medications, when used with prednisolone, were superior to prednisolone alone in a multi-centre randomized trial of 139 patients with new-onset JDM140. Clinicians have the choice of oral prednisolone (12 mg/kg/day with ceiling doses applied, typically capping at 60 mg/day) or intravenous methylprednisolone (10–30 mg/kg/day, maximum of 1 g/day)126,133,134,139. Intravenous administration might result in an increased therapeutic effect and less toxicity than with an oral corticosteroid and should be considered, especially when there are concerns about gastrointestinal absorption126,133. Intravenous methylprednisolone might have the additional benefit of reducing skin disease more rapidly than oral prednisolone141.
Evidence is lacking to determine the best second-line treatment when the combination of corticosteroids and methotrexate does not adequately control disease or patients are intolerant to methotrexate. Head-to-head comparison studies are needed. In the absence of current evidence, CARRA have developed a series of consensus treatment plans to limit treatment variation among patients and enable comparative effectiveness studies from registry data134,135,136,142. Some evidence, in the form of case series involving small to moderate numbers of patients, supports the use of mycophenolate mofetil (MMF) for the treatment of skin or muscle disease143,144,145,146. Evidence for the use of azathioprine comes from historical studies that included small numbers of patients, and although this drug can be used as an adjunctive treatment, it has become less favoured over the last two decades for the treatment of IIM in paediatric practice147,148. Some evidence is available on the use of tacrolimus to treat JIIM but is limited by the small number of patients involved149,150,151. Adult data suggest that tacrolimus or ciclosporin alongside corticosteroids should be considered for patients with myositis-associated ILD, and although these data are often extrapolated to JIIM, insufficient data are available to form evidence-based recommendations for this complication in childhood-onset disease133. Data from case series of adult and paediatric patients suggest that cyclophosphamide or rituximab could be considered when ILD is present and should be used early, potentially as part of an induction regime133. Although no standardized treatment guidelines are available on the management of ILD in adult patients with IIM, a summary of evidence and treatment approach has been presented in a review and, in the absence of evidence in JIIM, might provide useful guidance in the treatment approaches for childhood-onset disease152. In this review, the authors suggests that corticosteroids are used as the initial treatment for acute disease followed by MMF or azathioprine as first-line steroid-sparing agents. Tacrolimus is suggested as an appropriate second-line steroid-sparing agent for patients with disease that is refractory to MMF or azathioprine, or for select patients with severe disease. Cyclophosphamide is proposed as a third-line steroid-sparing agent. IVIG or rituximab are advocated as appropriate adjunctive agents in combination with traditional steroid-sparing agents for patients with refractory disease152.
Intravenous immunoglobulin (IVIG) might be a helpful adjunct for severe or refractory skin disease, muscle inflammation or dysphagia133. In a randomized placebo-controlled 16-week trial of IVIG in adult patients with dermatomyositis (the ProDERM trial), a higher proportion of patients in the IVIG treatment group reached the primary outcome of total improvement score (a composite measure of disease activity) of at least 20 (indicating at least minimal improvement) than in the placebo control group (P < 0.001)153. Although evidence in adult-onset disease includes randomized trials, evidence in JIIM is mostly limited to cohort studies or case series154,155,156,157,158,159. Interpreting observational evidence is challenging owing to the use of concomitant therapies, the variable doses or treatment courses of IVIG used and the small numbers of patients involved. In one notable study, the researchers applied bias reduction methods to assess the efficacy of IVIG in a retrospective cohort of 78 patients with JDM, demonstrating that IVIG was efficacious in controlling severe or refractory disease, particularly in those patients who had steroid-resistant disease158. Other immunomodulating drugs have also been reported to improve symptoms of dysphagia or improve objective measures of swallowing function133. Cyclophosphamide tends to be reserved for more severe or refractory disease in view of the toxicity of this drug, but might be considered in cases of major organ involvement, including ILD or ulcerative skin disease126,133,157,158,160,161. Despite a lack of evidence from randomized controlled trials, the use of IVIG or cyclophosphamide is supported by case reports, case series and analysis by marginal structural modelling157,158,159,160.
Evidence related to the treatment of skin manifestations in JIIM is limited, but IVIG or rituximab can be used to treat skin manifestations refractory to corticosteroid and DMARDs133,157,158. In the ProDERM trial, IVIG was efficacious in improving skin disease activity in patients with adult-onset dermatomyositis, as measured by the modified Cutaneous Dermatomyositis Disease Severity and Activity Index153. Despite the relative lack of evidence for use of hydroxychloroquine in JIIM, limited to case series with small numbers of patients, this drug is often used as an adjunctive treatment for skin disease and arthritis162,163,164. Hydroxychloroquine is included in the CARRA Consensus Treatment Plan for skin predominant disease142. However, in a prospective study of 184 children with JDM treated at a single children’s hospital, although hydroxychloroquine was often administered to those patients with higher skin activity scores, the drug did not lead to any statistically significant improvement in skin rash by the end of the observation period141. Topical tacrolimus (0.1%) or topical corticosteroids might help localized skin disease, particularly for symptomatic redness or itching126.
The use of biologics in JIIM has been summarized elsewhere in a systematic review165. Rituximab treatment for refractory muscle or skin disease is supported by one randomized controlled trial and various case series or cohort studies108,166,167,168,169,170. In the Rituximab in Myositis randomized controlled trial, despite failure to meet the primary or secondary endpoints, 83% of the patients met the definition of improvement108. Data were reported in aggregate but post hoc analyses suggested that patients with JIIM were more likely to respond to treatment than those patients with adult-onset myositis133,166. The presence of anti-Mi2 antibodies, anti-synthetase antibodies or other undefined autoantibodies were other predictors of a beneficial response, but anti-Mi2 antibodies and anti-synthetase antibodies are less common in JIIM than in adult disease165,166,168. In this trial, rituximab treatment also led to improvement in cutaneous disease168. Evidence in adult-onset myositis (that is, data from retrospective and prospective studies rather than randomized controlled trial data) suggests that rituximab might be helpful in IIM-related ILD, but more data are needed in JIIM64,133,152.
Data from case series and cohort studies suggest that TNF blockade by infliximab or adalimumab can be helpful for refractory muscle or skin disease, including calcinosis165,171,172,173,174,175. In an open-label 12-week trial of the TNF inhibitor etanercept, the drug showed no appreciable benefit in nine patients with refractory JDM174, whereas etanercept had a steroid-sparing effect in a randomized double-blind placebo-controlled 52-week trial involving 16 patients with adult-onset IIM176. In rare instances, TNF inhibitors have been reported to induce myositis or cause disease flares in adult patients with IIM177,178. Although TNF inhibitors, particularly adalimumab or infliximab, might be helpful in some patients with JIIM, evidence from a systemic review suggest that treatment with these drugs does not lead to complete remission and better treatments are needed165. Abatacept has demonstrated efficacy in a randomized controlled trial in adult onset myositis179 and in an open label therapeutic trial in JIIM180. Abatacept might be helpful for the treatment of resistant disease, including calcinosis179,181,182.
JAK–STAT inhibitors target the interferon pathway and show clear promise in the treatment of IIM-related muscle, skin and lung disease7,64,183. A number of reports have highlighted the potential safety and efficacy of JAK inhibitors (including tofacitinib and baricitinib) in treatment-resistant adult IIM7,184. In JIIM, various JAK inhibitors (including baricitinib, tofacitinib and ruxolitinib) have shown promise in numerous case reports and case series, predominantly involving patients with refractory muscle or skin disease that is unresponsive to alternative immunosuppressive treatment(s)8,9,10,185,186,187. These studies have been carefully reviewed in a systematic review elsewhere, which described 48 publications reporting 145 unique patients (including 61 cases of JIIM) with refractory disease at baseline and demonstrated that treatment with JAK inhibitors led to improvement in a wide range of manifestations, including of the skin, muscle and lungs7. As well as providing evidence on the clinical efficacy of JAK inhibition, these studies suggest that JAK inhibitors can modulate the disease at an immunopathogenic level, as demonstrated by the downregulation of interferon biomarkers, the type I interferon signature and STAT1 phosphorylation in T cells and monocytes to similar levels to that in healthy individuals8,9,10,11,186. These encouraging results suggest that JAK inhibition could be an effective, targeted treatment for JDM, and highlight the importance of confirming these findings in clinical trials7,183,188.
An important challenge in JIIM is the treatment of calcinosis. Some evidence is available on the use of DMARDs, medications that affect calcium and phosphorus metabolism, mechanical therapies and adjunctive therapies in the treatment of calcinosis in JIIM, as reviewed elsewhere189,190,191. However, the available evidence is limited and largely based on case reports or case series, cohort studies or limited controlled studies. A major unmet need exists for an improved understanding of calcinosis pathogenesis, for standardized tools to measure calcinosis and for efficacious treatment of this burdensome complication189,190. Consensus guidelines advocate for early aggressive treatment at disease onset to decrease the long-term risk of calcinosis, as well as consideration of an early increase in treatment of ongoing disease activity and intensifying immunosuppressive therapy in the presence of calcinosis126,133. Other than associations with some MSAs, as described above, evidence on risk factors for calcinosis is limited, but a single-centre retrospective study of 172 patients identified nailfold capillary abnormalities at baseline as a risk factor for calcinosis in univariate and multivariate analysis192. Some data are available on the histopathological and chemical composition of calcinosis, genetic and inflammatory markers in IIM-associated calcinosis and potential biomarkers of this complication, which have been reviewed in further detail elsewhere193.
Exercise
Cardiorespiratory fitness can be impaired in patients with JIIM during both inactive and active disease and in patients with both monocyclic and polycyclic disease courses owing to factors such as cardiovascular deconditioning and reduced thoracic compliance194,195,196,197,198. Studies, including a randomized controlled trial in children and adolescents with JDM, have demonstrated the safety and efficacy of exercise training programmes, including the positive effects of these programmes on health-related quality of life199,200,201. Hence, the management of JIIM should include a safe and appropriate exercise programme that is led and monitored by a specialist physiotherapist and/or occupational therapist126,133.
Some data are available on the efficacy of interventions to reduce fatigue in paediatric conditions such as JIIM, including land or aquatic-based exercise, medications and psychological interventions, which have been evaluated in a systematic review elsewhere202; however, in this study, the efficacy of current interventions to reduce fatigue could not be established owing to insufficient evidence. Fatigue is multi-dimensional and does not necessarily always correlate with disease activity and is instead strongly associated with biological, lifestyle, psychological and social factors202. Further multidimensional intervention studies are needed to identify the best management of this troublesome symptom.
Psychological support
JIIM has a notable impact on the emotional health of young people and their families203,204 Mental health issues, most commonly anxiety and depression, are reported frequently by children and young people with JIIM204,205. Psychological wellbeing, psychiatric comorbidities and health-related quality of life should be assessed using age-appropriate tools133. Access to mental health provision, ideally embedded within paediatric rheumatology services so that young people feel that counsellors understand their disease, is paramount204,205,206. Factors that impact negatively on the health-related quality of life of patients, including pain, muscle weakness, functional impairment or physical disability, poor sleep and fatigue, should be managed appropriately25,133,207,208.
Assessment of disease activity and treatment response
Disease activity should be measured in a quantifiable way in both clinical practice and clinical research studies to determine changes in disease activity over time and response to treatment. Tools to measure disease activity have been comprehensively reviewed by others129,209. The International Myositis and Clinical Studies group and Paediatric Rheumatology International Trials Organisation (PRINTO) have developed core sets to measure disease activity and damage, predominantly for use in research studies or clinical trials129,209. The ability to robustly define response to therapy is crucial for conducting clinical trials and has been addressed by the development of ACR–EULAR response criteria210,211. To define the optimal set of items collected in clinical practice to enable entry into research registries and comparison of data over time, an international collaboration has defined a consensus core dataset that is in use by several major registry studies15,212,213,214. Consensus recommendations advise the routine use of measures such as the Manual Muscle Testing in eight muscle groups (MMT-8) tool and the Childhood Myositis Assessment Scale (CMAS) to assess muscle strength and function126. Age-specific considerations need to be taken into account when using tools that measure muscle strength, function and quality of life133. For example, for the CMAS, results for head-lift, leg-lift and sit-up manoeuvres are dependent on the age of the patient and very young children should not be expected to achieve a score of 52, even when disease is inactive215,216. A shortened version of the MMT-8 tool that tests four (MMT-4) or six (MMT-6) muscle groups and a hybrid version that includes all eight items of the MMT-8 tool and three items from the CMAS have demonstrated good measurement properties and might be more suitable than MMT-8 or CMAS for routine clinical use217,218. More work is needed to define and reach consensus on the optimal tools for assessment of skin disease activity and measurement of quality of life in JDM129,214. Several tools are currently available, including the Cutaneous Assessment Tool, Disease Activity Score and Myositis Intention to Treat Activity Index, each of which correlates with the physicians’; skin Visual Analogue Scale219; furthermore, the Cutaneous Disease Area and Severity Index has been extensively used in studies of adult dermatomyositis and might be equally valuable for use in JDM129,209,220.
The importance of patient-reported outcome measures in outcome assessment within trials and in the clinic is becoming increasingly recognized. A study that included patients with JDM suggested that three tools from the Patient-Reported Outcomes Measurement Information System are an improvement over the previously widely-used Childhood Health Assessment Questionnaire (CHAQ) for capturing patient-reported outcomes221. Patient-Reported Outcomes Measurement Information System tools can be administered as fixed short forms or via computerized adaptive testing, the latter of which results in less pronounced floor and/or ceiling effects than fixed short forms222.
Conclusions
Our understanding and management of juvenile onset myositis has changed considerably in the past two decades, but numerous challenges remain (Box 1) and much work is still needed. A deeper appreciation of the underlying mechanisms that initiate and perpetuate inflammation of the blood vessels, muscles, skin and other organs, and how inflammatory mechanisms intersect with other aetiological pathways in JIIM, is needed. New insights are becoming available from studies at a single-cell level of gene expression, function and metabolic profiles. Further studies on the mechanisms of important patient-reported symptoms, such as fatigue, are also much needed. A vital aim is to ensure that novel data on underlying mechanisms are shared collaboratively and made accessible to drive the design of biomarker studies and enable validation studies and meta-analyses. The highly collaborative nature of myositis research, both basic and clinical, has enabled major progress thus far, and will support such platforms through which to generate evidence for new treatments, despite the rarity of JIIM (Box 1).
This strongly collaborative community, across paediatric, adolescent and adult myositis research, is reflected in the first ‘age-inclusive’ trial in myositis (the Rituximab in Myositis trial)108; such a design enables faster results for children and young people, rather than waiting for a ‘child-specific’ trial for drugs that have been granted a licence in adults. Progress has also been made using longitudinal observational data to support so-called ‘trial in silico’ analyses223; this approach will become more possible through widespread use of an agreed common clinical dataset214, which is embedded in clinical care and large research registries15,212,213. Translation of this core dataset for use in adolescent and adult care could facilitate evidence generation on long-term outcomes, which is currently lacking. Current long-term outcome data clearly indicate increased risks of cardiovascular or pulmonary disease in patients with IIMs compared with the general population. In the future, the integration of biomarker and pathogenesis data with long-term outcome data of those treated in the modern era will be critical for informing our patients and their families about comorbidities, outcomes and the chances of sustained remission.
Ultimately, a combination of a better understanding of disease mechanisms, biomarkers that accurately track disease activity, including subclinical disease, and definitions of outcomes that include the patient perspective will be needed to deliver a personalized approach to managing myositis in children, and in the young people and adults they become.
References
Lundberg, I. E. et al. European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Arthritis Rheumatol. 69, 2271–2282 (2017).
Patel, B., Khan, N. & Werth, V. P. Applicability of EULAR/ACR classification criteria for dermatomyositis to amyopathic disease. J. Am. Acad. Dermatol. 79, 77–83.e1 (2018).
Bohan, A. & Peter, J. B. Polymyositis and dermatomyositis (second of two parts). N. Engl. J. Med. 292, 403–407 (1975).
Tansley, S. L. et al. Autoantibodies in juvenile-onset myositis: their diagnostic value and associated clinical phenotype in a large UK cohort. J. Autoimmun. 84, 55–64 (2017).
Rider, L. G. et al. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine 92, 223–243 (2013).
Papadopoulou, C. et al. The vasculopathy of juvenile dermatomyositis: endothelial injury, hypercoagulability, and increased arterial stiffness. Arthritis Rheumatol. 73, 1253–1266 (2021).
Paik, J. J. et al. Use of Janus kinase inhibitors in dermatomyositis: a systematic literature review. Clin. Exp. Rheumatol. 41, 348–358 (2023).
Sabbagh, S. et al. Treatment of anti-MDA5 autoantibody-positive juvenile dermatomyositis using tofacitinib. Brain 142, e59 (2019).
Aeschlimann, F. A. et al. A child with severe juvenile dermatomyositis treated with ruxolitinib. Brain 141, e80 (2018).
Papadopoulou, C., Hong, Y., Omoyinmi, E., Brogan, P. A. & Eleftheriou, D. Janus kinase 1/2 inhibition with baricitinib in the treatment of juvenile dermatomyositis. Brain 142, e8 (2019).
Ding, Y. et al. Janus kinase inhibitor significantly improved rash and muscle strength in juvenile dermatomyositis. Ann. Rheum. Dis. 80, 543–545 (2021).
Tansley, S. & Wedderburn, L. R. Comparing and contrasting clinical and serological features of juvenile and adult-onset myositis: implications for pathogenesis and outcomes. Curr. Opin. Rheumatol. 27, 601–607 (2015).
Loarce-Martos, J. et al. Clinical characteristics of juvenile idiopathic inflammatory myopathy and comparison with adult patients: analysis from a multicentric cohort in Spain. J. Clin. Rheumatol. 28, e195–e202 (2022).
Meyer, A. et al. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology 54, 50–63 (2015).
Martin, N. et al. A national registry for juvenile dermatomyositis and other paediatric idiopathic inflammatory myopathies: 10 years’ experience; the Juvenile Dermatomyositis National (UK and Ireland) Cohort biomarker study and repository for idiopathic inflammatory myopathies. Rheumatology 50, 137–145 (2011).
Ravelli, A. et al. Long-term outcome and prognostic factors of juvenile dermatomyositis: a multinational, multicenter study of 490 patients. Arthritis Care Res. 62, 63–72 (2010).
Huber, A. M. et al. Medium- and long-term functional outcomes in a multicenter cohort of children with juvenile dermatomyositis. Arthritis Rheum. 43, 541–549 (2000).
Okong’o, L. O., Esser, M., Wilmshurst, J. & Scott, C. Characteristics and outcome of children with juvenile dermatomyositis in Cape Town: a cross-sectional study. Pediatr. Rheumatol. Online J. 14, 60 (2016).
Sanner, H., Gran, J. T., Sjaastad, I. & Flato, B. Cumulative organ damage and prognostic factors in juvenile dermatomyositis: a cross-sectional study median 16.8 years after symptom onset. Rheumatology 48, 1541–1547 (2009).
Sharma, A., Gupta, A., Rawat, A., Suri, D. & Singh, S. Long-term outcome in children with juvenile dermatomyositis: a single-center study from North India. Int. J. Rheum. Dis. 23, 392–396 (2020).
Huber, A. M. et al. Early illness features associated with mortality in the juvenile idiopathic inflammatory myopathies. Arthritis Care Res. 66, 732–740 (2014).
Tsaltskan, V. et al. Long-term outcomes in juvenile myositis patients. Semin. Arthritis Rheum. 50, 149–155 (2020).
Rider, L. G. et al. Damage extent and predictors in adult and juvenile dermatomyositis and polymyositis as determined with the myositis damage index. Arthritis Rheum. 60, 3425–3435 (2009).
Mathiesen, P. et al. Long-term outcome in patients with juvenile dermatomyositis: a cross-sectional follow-up study. Scand. J. Rheumatol. 41, 50–58 (2012).
Apaz, M. T. et al. Health-related quality of life of patients with juvenile dermatomyositis: results from the Pediatric Rheumatology International Trials Organisation multinational quality of life cohort study. Arthritis Rheum. 61, 509–517 (2009).
Nordal, E. et al. Growth and puberty in juvenile dermatomyositis: a longitudinal cohort study. Arthritis Care Res. 72, 265–273 (2020).
Boros, C. et al. Juvenile dermatomyositis: what comes next? Long-term outcomes in childhood myositis from a patient perspective. Pediatr. Rheumatol. Online J. 20, 102 (2022).
Silverberg, J. I., Kwa, L., Kwa, M. C., Laumann, A. E. & Ardalan, K. Cardiovascular and cerebrovascular comorbidities of juvenile dermatomyositis in US children: an analysis of the national inpatient sample. Rheumatology 57, 694–702 (2018).
Coyle, K. et al. Metabolic abnormalities and cardiovascular risk factors in children with myositis. J. Pediatr. 155, 882–887 (2009).
Schwartz, T. et al. In active juvenile dermatomyositis, elevated eotaxin and MCP-1 and cholesterol levels in the upper normal range are associated with cardiac dysfunction. Rheumatology 53, 2214–2222 (2014).
Witczak, B. N. et al. Associations between cardiac and pulmonary involvement in patients with juvenile dermatomyositis — a cross-sectional study. Rheumatol. Int. 42, 1213–1220 (2022).
Shah, M. et al. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine 92, 25–41 (2013).
Walling, H. W., Gerami, P. & Sontheimer, R. D. Juvenile-onset clinically amyopathic dermatomyositis: an overview of recent progress in diagnosis and management. Paediatr. Drugs 12, 23–34 (2010).
Mamyrova, G. et al. Features distinguishing clinically amyopathic juvenile dermatomyositis from juvenile dermatomyositis. Rheumatology 57, 1956–1963 (2018).
McCann, L. J., Li, C. K., Varsani, H., Wedderburn, L. R. & Pilkington, C. A. Failure to over express MHC-CLASS-1 on muscle biopsy in a case of amyopathic juvenile dermatomyositis. Clin. Exp. Rheumatol. 25, 96–98 (2007).
Dalakas, M. C. & Hohlfeld, R. Polymyositis and dermatomyositis. Lancet 362, 971–982 (2003).
Kishi, T. et al. Association of anti-3-Hydroxy-3-Methylglutaryl-Coenzyme A reductase autoantibodies with DRB1*07:01 and severe myositis in juvenile myositis patients. Arthritis Care Res. 69, 1088–1094 (2017).
Li, D. & Tansley, S. L. Juvenile dermatomyositis — clinical phenotypes. Curr. Rheumatol. Rep. 21, 74 (2019).
Orandi, A. B. et al. Assessment, classification and treatment of calcinosis as a complication of juvenile dermatomyositis: a survey of pediatric rheumatologists by the childhood arthritis and rheumatology research alliance (CARRA). Pediatr. Rheumatol. Online J. 15, 71 (2017).
Rider, L. G. & Nistala, K. The juvenile idiopathic inflammatory myopathies: pathogenesis, clinical and autoantibody phenotypes, and outcomes. J. Intern. Med. 280, 24–38 (2016).
Kwiatkowska, D. & Reich, A. The significance of autoantibodies in juvenile dermatomyositis. Biomed. Res. Int. 2021, 5513544 (2021).
Best, M. et al. Use of anti-transcriptional intermediary factor-1 γ autoantibody in identifying adult dermatomyositis patients with cancer: a systematic review and meta-analysis. Acta Derm. Venereol. 99, 256–262 (2019).
Oldroyd, A. G. S. et al. A systematic review and meta-analysis to inform cancer screening guidelines in idiopathic inflammatory myopathies. Rheumatology 60, 2615–2628 (2021).
Gunawardena, H. et al. Autoantibodies to a 140-kd protein in juvenile dermatomyositis are associated with calcinosis. Arthritis Rheum. 60, 1807–1814 (2009).
Espada, G., Maldonado Cocco, J. A., Fertig, N. & Oddis, C. V. Clinical and serologic characterization of an Argentine pediatric myositis cohort: identification of a novel autoantibody (anti-MJ) to a 142-kDa protein. J. Rheumatol. 36, 2547–2551 (2009).
Tansley, S. L. et al. Calcinosis in juvenile dermatomyositis is influenced by both anti-NXP2 autoantibody status and age at disease onset. Rheumatology 53, 2204–2208 (2014).
Rogers, A., Chung, L., Li, S., Casciola-Rosen, L. & Fiorentino, D. F. Cutaneous and systemic findings associated with nuclear matrix protein 2 antibodies in adult dermatomyositis patients. Arthritis Care Res. 69, 1909–1914 (2017).
Deakin, C. T. et al. Muscle biopsy findings in combination with myositis-specific autoantibodies aid prediction of outcomes in juvenile dermatomyositis. Arthritis Rheumatol. 68, 2806–2816 (2016).
Yamasaki, Y. et al. Clinical impact of myositis-specific autoantibodies on long-term prognosis of juvenile idiopathic inflammatory myopathies: multicentre study. Rheumatology 60, 4821–4831 (2021).
Mamyrova, G. et al. Anti-MDA5 autoantibodies associated with juvenile dermatomyositis constitute a distinct phenotype in North America. Rheumatology 60, 1839–49 (2021).
Kobayashi, N. et al. Clinical and laboratory features of fatal rapidly progressive interstitial lung disease associated with juvenile dermatomyositis. Rheumatology 54, 784–791 (2015).
Tansley, S. L. et al. Anti-MDA5 autoantibodies in juvenile dermatomyositis identify a distinct clinical phenotype: a prospective cohort study. Arthritis Res. Ther. 16, R138 (2014).
Fiorentino, D., Chung, L., Zwerner, J., Rosen, A. & Casciola-Rosen, L. The mucocutaneous and systemic phenotype of dermatomyositis patients with antibodies to MDA5 (CADM-140): a retrospective study. J. Am. Acad. Dermatol. 65, 25–34 (2011).
Melki, I. et al. Anti-MDA5 juvenile idiopathic inflammatory myopathy: a specific subgroup defined by differentially enhanced interferon-α signalling. Rheumatology 59, 1927–1937 (2020).
Kobayashi, I. et al. Anti-melanoma differentiation-associated gene 5 antibody is a diagnostic and predictive marker for interstitial lung diseases associated with juvenile dermatomyositis. J. Pediatr. 158, 675–677 (2011).
Ueki, M. et al. Myositis-specific autoantibodies in Japanese patients with juvenile idiopathic inflammatory myopathies. Mod. Rheumatol. 29, 351–356 (2019).
Nombel, A., Fabien, N. & Coutant, F. Dermatomyositis with anti-MDA5 antibodies: bioclinical features, pathogenesis and emerging therapies. Front. Immunol. 12, 773352 (2021).
Yasin, S. A. et al. Histological heterogeneity in a large clinical cohort of juvenile idiopathic inflammatory myopathy: analysis by myositis autoantibody and pathological features. Neuropathol. Appl. Neurobiol. 45, 495–512 (2019).
Caproni, M. et al. Amyopathic dermatomyositis: a review by the Italian Group of Immunodermatology. Arch. Dermatol. 138, 23–27 (2002).
Betteridge, Z. E. et al. Clinical and human leucocyte antigen class II haplotype associations of autoantibodies to small ubiquitin-like modifier enzyme, a dermatomyositis-specific autoantigen target, in UK Caucasian adult-onset myositis. Ann. Rheum. Dis. 68, 1621–1625 (2009).
Kishi, T. et al. Anti-SAE autoantibody-positive Japanese patient with juvenile dermatomyositis complicated with interstitial lung disease — a case report. Pediatr. Rheumatol. Online J. 19, 34 (2021).
Abe, S. et al. Clinically amyopathic dermatomyositis associated with anti-nuclear matrix protein 2 antibody. Rheumatol. Adv. Pract. 5, rkab104 (2021).
Damoiseaux, J., Mammen, A. L., Piette, Y., Benveniste, O. & Allenbach, Y. 256th ENMC international workshop: myositis specific and associated autoantibodies (MSA-ab): Amsterdam, The Netherlands, 8–10 October 2021. Neuromuscul. Disord. 32, 594–608 (2022).
Lundberg, I. E. et al. Idiopathic inflammatory myopathies. Nat. Rev. Dis. Prim. 7, 86 (2021).
Martins, P. et al. Clinical characterisation of a multicentre nationwide cohort of patients with antisynthetase syndrome. ARP Rheumatol. 1, 190–196 (2022).
Pinal-Fernandez, I., Casal-Dominguez, M. & Mammen, A. L. Immune-mediated necrotizing myopathy. Curr. Rheumatol. Rep. 20, 21 (2018).
Binns, E. L. et al. Effective induction therapy for anti-SRP associated myositis in childhood: a small case series and review of the literature. Pediatr. Rheumatol. Online J. 15, 77 (2017).
Allenbach, Y. & Benveniste, O. Peculiar clinicopathological features of immune-mediated necrotizing myopathies. Curr. Opin. Rheumatol. 30, 655–663 (2018).
Tansley, S. L. et al. Anti-HMGCR autoantibodies in Juvenile idiopathic inflammatory myopathies identify a rare but clinically important subset of patients. J. Rheumatol. 44, 488–492 (2017).
Mamyrova, G. et al. Clinical and laboratory features distinguishing juvenile polymyositis and muscular dystrophy. Arthritis Care Res. 65, 1969–1975 (2013).
Wedderburn, L. R. et al. HLA class II haplotype and autoantibody associations in children with juvenile dermatomyositis and juvenile dermatomyositis-scleroderma overlap. Rheumatology 46, 1786–1791 (2007).
Khaosut, P., Pilkington, C., Wedderburn, L. R. & Compeyrot-Lacassagne, S. An international survey of developing classification criteria for juvenile dermatomyositis-scleroderma overlap. Rheumatology 58, 2062–2064 (2019).
Bitencourt, N., Solow, E. B., Wright, T. & Bermas, B. L. Inflammatory myositis in systemic lupus erythematosus. Lupus 29, 776–781 (2020).
Sabbagh, S. et al. Anti-Ro52 autoantibodies are associated with interstitial lung disease and more severe disease in patients with juvenile myositis. Ann. Rheum. Dis. 78, 988–995 (2019).
Orandi, A. B., Eutsler, E., Ferguson, C., White, A. J. & Kitcharoensakkul, M. Sarcoidosis presenting as granulomatous myositis in a 16-year-old adolescent. Pediatr. Rheumatol. Online J. 14, 59 (2016).
Kang, Y. et al. Muscle involvement in Polyarteritis nodosa: report of eight cases with characteristic contrast enhancement pattern on MRI. AJR Am. J. Roentgenol. 206, 378–384 (2016).
Rama, M. et al. A decision tree for the genetic diagnosis of deficiency of adenosine deaminase 2 (DADA2): a French reference centres experience. Eur. J. Hum. Genet 26, 960–971 (2018).
Eleftheriou, D. & Brogan, P. A. Genetic interferonopathies: an overview. Best. Pract. Res. Clin. Rheumatol. 31, 441–459 (2017).
Kim, H. et al. Expression of interferon-regulated genes in juvenile dermatomyositis versus Mendelian autoinflammatory interferonopathies. Arthritis Res. Ther. 22, 69 (2020).
Fujikawa, K., Migita, K., Tsukada, T., Kawakami, A. & Eguchi, K. Protracted febrile myalgia syndrome in a Japanese patient with fasciitis detected on MRI. Intern. Med. 53, 2817–2819 (2014).
Wedderburn, L. R. & Rider, L. G. Juvenile dermatomyositis: new developments in pathogenesis, assessment and treatment. Best. Pract. Res. Clin. Rheumatol. 23, 665–678 (2009).
Mamyrova, G., Rider, L. G., Haagenson, L., Wong, S. & Brown, K. E. Parvovirus B19 and onset of juvenile dermatomyositis. J. Am. Med. Assoc. 294, 2170–2171 (2005).
Pachman, L. M. et al. Lack of detection of enteroviral RNA or bacterial DNA in magnetic resonance imaging-directed muscle biopsies from twenty children with active untreated juvenile dermatomyositis. Arthritis Rheum. 38, 1513–1518 (1995).
Pachman, L. M. et al. History of infection before the onset of juvenile dermatomyositis: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Research Registry. Arthritis Rheum. 53, 166–172 (2005).
Pachman, L. M., Nolan, B. E., DeRanieri, D. & Khojah, A. M. Juvenile dermatomyositis: new clues to diagnosis and therapy. Curr. Treatm Opt. Rheumatol. 7, 39–62 (2021).
Okada, Y. et al. Anti-NXP2 antibody-positive dermatomyositis developed after COVID-19 manifesting as type I interferonopathy. Rheumatology 61, e90–e92 (2022).
Islam, B., Ahmed, M., Islam, Z. & Begum, S. M. Severe acute myopathy following SARS-CoV-2 infection: a case report and review of recent literature. Skelet. Muscle 11, 10 (2021).
Okada, S. et al. Global surface ultraviolet radiation intensity may modulate the clinical and immunologic expression of autoimmune muscle disease. Arthritis Rheum. 48, 2285–2293 (2003).
Shah, M. et al. Brief report: ultraviolet radiation exposure is associated with clinical and autoantibody phenotypes in juvenile myositis. Arthritis Rheum. 65, 1934–1941 (2013).
Neely, J., Long, C. S., Sturrock, H., Kim, S. & Childhood, A. Rheumatology Research Alliance Registry I. Association of short-term ultraviolet radiation exposure and disease severity in juvenile dermatomyositis: results from the childhood arthritis and rheumatology research alliance legacy registry. Arthritis Care Res. 71, 1600–1605 (2019).
Orione, M. A. et al. Risk factors for juvenile dermatomyositis: exposure to tobacco and air pollutants during pregnancy. Arthritis Care Res. 66, 1571–1575 (2014).
Scalabrini, J. C. et al. Environmental factors associated with juvenile idiopathic inflammatory myopathy clinical and serologic phenotypes. Pediatr. Rheumatol. Online J. 20, 28 (2022).
Miller, F. W. et al. Genome-wide association study identifies HLA 8.1 ancestral haplotype alleles as major genetic risk factors for myositis phenotypes. Genes Immun. 16, 470–480 (2015).
Rothwell, S. et al. Dense genotyping of immune-related loci in idiopathic inflammatory myopathies confirms HLA alleles as the strongest genetic risk factor and suggests different genetic background for major clinical subgroups. Ann. Rheum. Dis. 75, 1558–1566 (2016).
Deakin, C. T. et al. Association with HLA-DRβ1 position 37 distinguishes juvenile dermatomyositis from adult-onset myositis. Hum. Mol. Genet. 31, 2471–2481 (2022).
Rothwell, S. et al. Focused HLA analysis in Caucasians with myositis identifies significant associations with autoantibody subgroups. Ann. Rheum. Dis. 78, 996–1002 (2019).
Chinoy, H. et al. The protein tyrosine phosphatase N22 gene is associated with juvenile and adult idiopathic inflammatory myopathy independent of the HLA 8.1 haplotype in British Caucasian patients. Arthritis Rheum. 58, 3247–3254 (2008).
Jani, M. et al. Genotyping of immune-related genetic variants identifies TYK2 as a novel associated locus for idiopathic inflammatory myopathies. Ann. Rheum. Dis. 73, 1750–1752 (2014).
McLellan, K. & Papadopoulou, C. Update on biomarkers of vasculopathy in juvenile and adult myositis. Curr. Rheumatol. Rep. 24, 227–237 (2022).
Kishi, T. et al. Endothelial activation markers as disease activity and damage measures in juvenile dermatomyositis. J. Rheumatol. 47, 1011–1018 (2020).
Khojah, A. et al. Association of p155/140 autoantibody with loss of nailfold capillaries but not generalized lipodystrophy: a study of ninety-six children with juvenile dermatomyositis. Arthritis Care Res. 74, 1065–1069 (2022).
Bloom, B. J., Miller, L. C. & Blier, P. R. Soluble adhesion molecules in pediatric rheumatic diseases. J. Rheumatol. 29, 832–836 (2002).
Soponkanaporn, S. et al. Expression of myxovirus-resistance protein A: a possible marker of muscle disease activity and autoantibody specificities in juvenile dermatomyositis. Neuropathol. Appl. Neurobiol. 45, 410–420 (2019).
Rodero, M. P. et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J. Exp. Med. 214, 1547–1555 (2017).
Roberson, E. D. O. et al. Transcriptomes of peripheral blood mononuclear cells from juvenile dermatomyositis patients show elevated inflammation even when clinically inactive. Sci. Rep. 12, 275 (2022).
Moneta, G. M. et al. Muscle expression of type I and type II interferons is increased in juvenile dermatomyositis and related to clinical and histologic features. Arthritis Rheumatol. 71, 1011–1021 (2019).
Platanias, L. C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 (2005).
Oddis, C. V. et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum. 65, 314–324 (2013).
Morita, R. et al. Human blood CXCR5+CD4+ T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34, 108–121 (2011).
Wilkinson, M. G. L. et al. Using peripheral blood immune signatures to stratify patients with adult and juvenile inflammatory myopathies. Rheumatology 59, 194–204 (2020).
Varsani, H. et al. Validation of a score tool for measurement of histological severity in juvenile dermatomyositis and association with clinical severity of disease. Ann. Rheum. Dis. 74, 204–210 (2015).
Nistala, K. et al. Myeloid related protein induces muscle derived inflammatory mediators in juvenile dermatomyositis. Arthritis Res. Ther. 15, R131 (2013).
Sag, E. et al. Inflammatory milieu of muscle biopsies in juvenile dermatomyositis. Rheumatol. Int. 41, 77–85 (2021).
Piper, C. J. M. et al. CD19+CD24hiCD38hi B cells are expanded in juvenile dermatomyositis and exhibit a pro-inflammatory phenotype after activation through toll-like receptor 7 and interferon-α. Front. Immunol. 9, 1372 (2018).
Turnier, J. L. et al. Comparison of lesional juvenile myositis and lupus skin reveals overlapping yet unique disease pathophysiology. Arthritis Rheumatol. 73, 1062–1072 (2021).
Brinkmann, V. et al. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004).
Duvvuri, B. et al. Neutrophil extracellular traps in tissue and periphery in juvenile dermatomyositis. Arthritis Rheumatol. 72, 348–358 (2020).
Seto, N. et al. Neutrophil dysregulation is pathogenic in idiopathic inflammatory myopathies. JCI Insight 5, e134189 (2020).
Caielli, S. et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med 213, 697–713 (2016).
Zhong, D. et al. Co-expression network analysis reveals the pivotal role of mitochondrial dysfunction and interferon signature in juvenile dermatomyositis. PeerJ 8, e8611 (2020).
Wilkinson, M. G. L. et al. Role of CD14+ monocyte-derived oxidised mitochondrial DNA in the inflammatory interferon type 1 signature in juvenile dermatomyositis. Ann. Rheum. Dis. https://doi.org/10.1136/ard-2022-223469 (2022).
Sabbagh, S. E. et al. Anti-mitochondrial autoantibodies are associated with cardiomyopathy, dysphagia, and features of more severe disease in adult-onset myositis. Clin. Rheumatol. 40, 4095–4100 (2021).
Bhattarai, S. et al. The immunoproteasomes are key to regulate myokines and MHC class I expression in idiopathic inflammatory myopathies. J. Autoimmun. 75, 118–129 (2016).
Nagaraju, K. et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 52, 1824–1835 (2005).
Ma, X. et al. Endoplasmic reticulum stress is involved in muscular pathogenesis in idiopathic inflammatory myopathies. Front. Cell Dev. Biol. 10, 791986 (2022).
Bellutti Enders, F. et al. Consensus-based recommendations for the management of juvenile dermatomyositis. Ann. Rheum. Dis. 76, 329–340 (2017).
Kobayashi, I. et al. Clinical practice guidance for juvenile dermatomyositis (JDM) 2018-update. Mod. Rheumatol. 30, 411–423 (2020).
Tansley, S. L. et al. The promise, perceptions, and pitfalls of immunoassays for autoantibody testing in myositis. Arthritis Res. Ther. 22, 117 (2020).
Kim, H., Huber, A. M. & Kim, S. Updates on juvenile dermatomyositis from the last decade: classification to outcomes. Rheum. Dis. Clin. North Am. 47, 669–690 (2021).
Tansley, S. L., Li, D., Betteridge, Z. E. & McHugh, N. J. The reliability of immunoassays to detect autoantibodies in patients with myositis is dependent on autoantibody specificity. Rheumatology 59, 2109–2114 (2020).
Satoh, M., Tanaka, S., Ceribelli, A., Calise, S. J. & Chan, E. K. A comprehensive overview on myositis-specific antibodies: new and old biomarkers in idiopathic inflammatory myopathy. Clin. Rev. Allergy Immunol. 52, 1–19 (2017).
Espinosa-Ortega, F. et al. Comparison of autoantibody specificities tested by a line blot assay and immunoprecipitation-based algorithm in patients with idiopathic inflammatory myopathies. Ann. Rheum. Dis. 78, 858–860 (2019).
Oldroyd, A. G. S. et al. British Society for Rheumatology guideline on management of paediatric, adolescent and adult patients with idiopathic inflammatory myopathy. Rheumatology 61, 1760–1768 (2022).
Huber, A. M. et al. Protocols for the initial treatment of moderately severe juvenile dermatomyositis: results of a Children’s Arthritis and Rheumatology Research Alliance Consensus Conference. Arthritis Care Res. 62, 219–225 (2010).
Huber, A. M. et al. Consensus treatments for moderate juvenile dermatomyositis: beyond the first two months. Results of the second Childhood Arthritis and Rheumatology Research Alliance consensus conference. Arthritis Care Res. 64, 546–553 (2012).
Huber, A. M. et al. Childhood Arthritis and Rheumatology Research Alliance Consensus Clinical Treatment Plans for juvenile dermatomyositis with persistent skin rash. J. Rheumatol. 44, 110–116 (2017).
McCann, L. J., Livermore, P., Wilkinson, M. G. L. & Wedderburn, L. R. Juvenile dermatomyositis. Where are we now? Clin. Exp. Rheumatol. 40, 394–403 (2022).
Wu, Q., Wedderburn, L. R. & McCann, L. J. Juvenile dermatomyositis: latest advances. Best. Pract. Res. Clin. Rheumatol. 31, 535–557 (2017).
Orandi, A. B. et al. Favorable outcomes with reduced steroid use in juvenile dermatomyositis. Pediatr. Rheumatol. Online J. 19, 127 (2021).
Ruperto, N. et al. Prednisone versus prednisone plus ciclosporin versus prednisone plus methotrexate in new-onset juvenile dermatomyositis: a randomised trial. Lancet 387, 671–678 (2016).
Wang, A., Morgan, G. A., Paller, A. S. & Pachman, L. M. Skin disease is more recalcitrant than muscle disease: a long-term prospective study of 184 children with juvenile dermatomyositis. J. Am. Acad. Dermatol. 84, 1610–1618 (2021).
Kim, S. et al. Childhood Arthritis and Rheumatology Research Alliance consensus clinical treatment plans for juvenile dermatomyositis with skin predominant disease. Pediatr. Rheumatol. Online J. 15, 1 (2017).
Rouster-Stevens, K. A., Morgan, G. A., Wang, D. & Pachman, L. M. Mycophenolate mofetil: a possible therapeutic agent for children with juvenile dermatomyositis. Arthritis Care Res. 62, 1446–1451 (2010).
Dagher, R. et al. Mycophenolate mofetil in juvenile dermatomyositis: a case series. Rheumatol. Int. 32, 711–716 (2012).
Cakan, M., Karadag, S. G. & Ayaz, N. A. Complete and sustained resolution of calcinosis universalis in a juvenile dermatomyositis case with mycophenolate mofetil. Turk. J. Pediatr. 61, 771–775 (2019).
Varnier, G. C. et al. Experience with the use of mycophenolate mofetil in juvenile idiopathic inflammatory myopathies. Rheumatology 62(SI2), SI163–SI169 (2022).
Jacobs, J. C. Methotrexate and azathioprine treatment of childhood dermatomyositis. Pediatrics 59, 212–218 (1977).
Ng, Y. T., Ouvrier, R. A. & Wu, T. Drug therapy in juvenile dermatomyositis: follow-up study. J. Child. Neurol. 13, 109–112 (1998).
Hassan, J., van der Net, J. J. & van Royen-Kerkhof, A. Treatment of refractory juvenile dermatomyositis with tacrolimus. Clin. Rheumatol. 27, 1469–1471 (2008).
Yamada, A., Ohshima, Y., Omata, N., Yasutomi, M. & Mayumi, M. Steroid-sparing effect of tacrolimus in a patient with juvenile dermatomyositis presenting poor bioavailability of cyclosporine A. Eur. J. Pediatr. 163, 561–562 (2004).
Rao, A. P., Kolli, V., Rajarathinam, I. & Raghuram, J. Tacrolimus in refractory juvenile dermatomyositis: case report and review of literature. Indian J. Rheumatol. 16, 205–208 (2020).
Hallowell, R. W. & Paik, J. J. Myositis-associated interstitial lung disease: a comprehensive approach to diagnosis and management. Clin. Exp. Rheumatol. 40, 373–383 (2022).
Aggarwal, R. et al. Trial of intravenous immune globulin in dermatomyositis. N. Engl. J. Med. 387, 1264–1278 (2022).
Dalakas, M. C. et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N. Engl. J. Med. 329, 1993–2000 (1993).
Miyasaka, N. et al. Effects of intravenous immunoglobulin therapy in Japanese patients with polymyositis and dermatomyositis resistant to corticosteroids: a randomized double-blind placebo-controlled trial. Mod. Rheumatol. 22, 382–393 (2012).
Aggarwal, R. et al. Prospective, double-blind, randomized, placebo-controlled phase III study evaluating efficacy and safety of octagam 10% in patients with dermatomyositis (“ProDERM Study”). Medicine 100, e23677 (2021).
Doudouliaki, T., Papadopoulou, C. & Deakin, C. T. Use of rescue therapy with IVIG or cyclophosphamide in juvenile myositis. Curr. Rheumatol. Rep. 23, 24 (2021).
Lam, C. G., Manlhiot, C., Pullenayegum, E. M. & Feldman, B. M. Efficacy of intravenous Ig therapy in juvenile dermatomyositis. Ann. Rheum. Dis. 70, 2089–2094 (2011).
Al-Mayouf, S. M., Laxer, R. M., Schneider, R., Silverman, E. D. & Feldman, B. M. Intravenous immunoglobulin therapy for juvenile dermatomyositis: efficacy and safety. J. Rheumatol. 27, 2498–2503 (2000).
Deakin, C. T. et al. Efficacy and safety of cyclophosphamide treatment in severe juvenile dermatomyositis shown by marginal structural modeling. Arthritis Rheumatol. 70, 785–793 (2018).
Riley, P. et al. Intravenous cyclophosphamide pulse therapy in juvenile dermatomyositis. A review of efficacy and safety. Rheumatology 43, 491–496 (2004).
Kishi, T. et al. Corticosteroid discontinuation, complete clinical response and remission in juvenile dermatomyositis. Rheumatology 60, 2134–2145 (2021).
Ang, G. C. & Werth, V. P. Combination antimalarials in the treatment of cutaneous dermatomyositis: a retrospective study. Arch. Dermatol. 141, 855–859 (2005).
Olson, N. Y. & Lindsley, C. B. Adjunctive use of hydroxychloroquine in childhood dermatomyositis. J. Rheumatol. 16, 1545–1547 (1989).
Marrani, E. et al. A systematic review on biological therapies in juvenile idiopathic inflammatory myopathies: an evidence gap in precision medicine. Clin. Exp. Rheumatol. 40, 457–470 (2022).
Aggarwal, R. et al. Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis. Arthritis Rheumatol. 66, 740–749 (2014).
Vargas Lebron, C., Ruiz Montesino, M. D., Moreira Navarrete, V. & Toyos Sainz de Miera, F. J. Treatment with rituximab in juvenile dermatomyositis: effect on calcinosis. Reumatol. Clin. 16, 368–370 (2020).
Aggarwal, R. et al. Cutaneous improvement in refractory adult and juvenile dermatomyositis after treatment with rituximab. Rheumatology 56, 247–254 (2017).
Bader-Meunier, B. et al. Safety and efficacy of rituximab in severe juvenile dermatomyositis: results from 9 patients from the French Autoimmunity and Rituximab registry. J. Rheumatol. 38, 1436–1440 (2011).
Cooper, M. A. et al. Rituximab for the treatment of juvenile dermatomyositis: a report of four pediatric patients. Arthritis Rheum. 56, 3107–3111 (2007).
Campanilho-Marques, R. et al. Retrospective analysis of infliximab and adalimumab treatment in a large cohort of juvenile dermatomyositis patients. Arthritis Res. Ther. 22, 79 (2020).
Riley, P. et al. Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology 47, 877–880 (2008).
Spencer, C. H. et al. Biologic therapies for refractory juvenile dermatomyositis: five years of experience of the Childhood Arthritis and Rheumatology Research Alliance in North America. Pediatr. Rheumatol. Online J. 15, 50 (2017).
Rouster-Stevens, K. A., Ferguson, L., Morgan, G., Huang, C. C. & Pachman, L. M. Pilot study of etanercept in patients with refractory juvenile dermatomyositis. Arthritis Care Res. 66, 783–787 (2014).
Green, K. L., Twilt, M. & Southwood, T. The role of etanercept in juvenile dermatomyositis (JDMS) in children. Pediatr. Rheumatol. Online J. 12, P279 (2014).
Muscle Study Group. A randomized, pilot trial of etanercept in dermatomyositis. Ann. Neurol. 70, 427–436 (2011).
Zengin, O. et al. Three cases of anti-TNF induced myositis and literature review. Rev. Bras. Reumatol. Engl. Ed. 57, 590–595 (2017).
Dastmalchi, M. et al. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann. Rheum. Dis. 67, 1670–1677 (2008).
Tjarnlund, A. et al. Abatacept in the treatment of adult dermatomyositis and polymyositis: a randomised, phase IIb treatment delayed-start trial. Ann. Rheum. Dis. 77, 55–62 (2018).
Curiel, R. V. et al. Improvement in disease activity in refractory juvenile dermatomyositis following abatacept therapy. Arthritis Rheumatol. https://doi.org/10.1002/art.42450 (2023).
Sukumaran, S. & Vijayan, V. Abatacept in the treatment of juvenile dermatomyositis-associated calcifications in a 16-year-old girl. Case Rep. Rheumatol. 2020, 4073879 (2020).
Arabshahi, B., Silverman, R. A., Jones, O. Y. & Rider, L. G. Abatacept and sodium thiosulfate for treatment of recalcitrant juvenile dermatomyositis complicated by ulceration and calcinosis. J. Pediatr. 160, 520–522 (2012).
Ll Wilkinson, M. G., Deakin, C. T., Papadopoulou, C., Eleftheriou, D. & Wedderburn, L. R. JAK inhibitors: a potential treatment for JDM in the context of the role of interferon-driven pathology. Pediatr. Rheumatol. Online J. 19, 146 (2021).
Chen, Z., Wang, X. & Ye, S. Tofacitinib in amyopathic dermatomyositis-associated interstitial lung disease. N. Engl. J. Med. 381, 291–293 (2019).
Le Voyer, T. et al. JAK inhibitors are effective in a subset of patients with juvenile dermatomyositis: a monocentric retrospective study. Rheumatology 60, 5801–5808 (2021).
Kim, H. et al. Janus kinase (JAK) inhibition with baricitinib in refractory juvenile dermatomyositis. Ann. Rheum. Dis. 80, 406–408 (2021).
Yu, Z., Wang, L., Quan, M., Zhang, T. & Song, H. Successful management with Janus kinase inhibitor tofacitinib in refractory juvenile dermatomyositis: a pilot study and literature review. Rheumatology 60, 1700–1707 (2021).
Kim, H. Updates on interferon in juvenile dermatomyositis: pathogenesis and therapy. Curr. Opin. Rheumatol. 33, 371–377 (2021).
Kul Cinar, O., Papadopoulou, C. & Pilkington, C. A. Treatment of calcinosis in juvenile dermatomyositis. Curr. Rheumatol. Rep. 23, 13 (2021).
Hoeltzel, M. F., Oberle, E. J., Robinson, A. B., Agarwal, A. & Rider, L. G. The presentation, assessment, pathogenesis, and treatment of calcinosis in juvenile dermatomyositis. Curr. Rheumatol. Rep. 16, 467 (2014).
Elahmar, H., Feldman, B. M. & Johnson, S. R. Management of calcinosis cutis in rheumatic diseases. J. Rheumatol. 49, 980–989 (2022).
Nozawa, T. et al. Early abnormal nailfold capillary changes are predictive of calcinosis development in juvenile dermatomyositis. J. Rheumatol. 49, 1250–1255 (2022).
Chung, M. P. et al. Calcinosis biomarkers in adult and juvenile dermatomyositis. Autoimmun. Rev. 19, 102533 (2020).
Berntsen, K. S. et al. Cardiorespiratory fitness in long-term juvenile dermatomyositis: a controlled, cross-sectional study of active/inactive disease. Rheumatology 58, 492–501 (2019).
Blom, K. J. et al. Trajectories of cardiorespiratory fitness in patients with juvenile dermatomyositis. Rheumatology 56, 2204–2211 (2017).
Hicks, J. E., Drinkard, B., Summers, R. M. & Rider, L. G. Decreased aerobic capacity in children with juvenile dermatomyositis. Arthritis Rheum. 47, 118–123 (2002).
Mathiesen, P. R. et al. Aerobic fitness after JDM — a long-term follow-up study. Rheumatology 52, 287–295 (2013).
Takken, T., Spermon, N., Helders, P. J., Prakken, A. B. & Van Der Net, J. Aerobic exercise capacity in patients with juvenile dermatomyositis. J. Rheumatol. 30, 1075–1080 (2003).
Habers, G. E. et al. Muscles in motion: a randomized controlled trial on the feasibility, safety and efficacy of an exercise training programme in children and adolescents with juvenile dermatomyositis. Rheumatology 55, 1251–1262 (2016).
Riisager, M., Mathiesen, P. R., Vissing, J., Preisler, N. & Orngreen, M. C. Aerobic training in persons who have recovered from juvenile dermatomyositis. Neuromuscul. Disord. 23, 962–968 (2013).
Astley, C. et al. Home-based exercise program for adolescents with juvenile dermatomyositis quarantined during COVID-19 pandemic: a mixed methods study. Pediatr. Rheumatol. Online J. 19, 159 (2021).
Kant-Smits, K., Van Brussel, M., Nijhof, S. & Van3 der Net, J. Reducing fatigue in ped33iatric rheumatic conditions: a systematic review. Pediatr. Rheumatol. Online J. 19, 111 (2021).
Livermore, P. et al. Being on the juvenile dermatomyositis rollercoaster: a qualitative study. Pediatr. Rheumatol. Online J. 17, 30 (2019).
Ardalan, K. et al. Parent perspectives on addressing emotional health for children and young adults with juvenile myositis. Arthritis Care Res. 73, 18–29 (2021).
Fawole, O. A. et al. Engaging patients and parents to improve mental health intervention for youth with rheumatological disease. Pediatr. Rheumatol. Online J. 19, 19 (2021).
Livermore, P. et al. Mapping the current psychology provision for children and young people with juvenile dermatomyositis. Rheumatol. Adv. Pract. 5, rkab062 (2021).
Butbul Aviel, Y. et al. Sleep and fatigue and the relationship to pain, disease activity and quality of life in juvenile idiopathic arthritis and juvenile dermatomyositis. Rheumatology 50, 2051–2060 (2011).
Tollisen, A., Sanner, H., Flato, B. & Wahl, A. K. Quality of life in adults with juvenile-onset dermatomyositis: a case-control study. Arthritis Care Res. 64, 1020–1027 (2012).
Rider, L. G. et al. Measures of adult and juvenile dermatomyositis, polymyositis, and inclusion body myositis: physician and patient/parent global activity, manual muscle testing (MMT), health assessment questionnaire (HAQ)/childhood health assessment questionnaire (C-HAQ), childhood myositis assessment scale (CMAS), myositis disease activity assessment tool (MDAAT), disease activity score (DAS), short form 36 (SF-36), child health questionnaire (CHQ), physician global damage, myositis damage index (MDI), quantitative muscle testing (QMT), myositis functional index-2 (FI-2), myositis activities profile (MAP), inclusion body myositis functional rating scale (IBMFRS), cutaneous dermatomyositis disease area and severity index (CDASI), cutaneous assessment tool (CAT), dermatomyositis skin severity index (DSSI), skindex, and dermatology life quality index (DLQI). Arthritis Care Res. 63, S118–S157 (2011).
Rider, L. G. et al. American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Juvenile Dermatomyositis: an International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 69, 911–923 (2017).
Aggarwal, R. et al. American College of Rheumatology/European League Against Rheumatism Criteria for Minimal, Moderate, and Major Clinical Response in Adult Dermatomyositis and Polymyositis: an International Myositis Assessment and Clinical Studies Group/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 69, 898–9130 (2017).
Liu, K. et al. Pilot study of the juvenile dermatomyositis consensus treatment plans: a CARRA registry study. J. Rheumatol. 48, 114–122 (2021).
Lilleker, J. B. et al. The EuroMyositis registry: an international collaborative tool to facilitate myositis research. Ann. Rheum. Dis. 77, 30–39 (2018).
McCann, L. J. et al. Development of a consensus core dataset in juvenile dermatomyositis for clinical use to inform research. Ann. Rheum. Dis. 77, 241–250 (2018).
Rennebohm, R. M. et al. Normal scores for nine maneuvers of the Childhood Myositis Assessment Scale. Arthritis Rheum. 51, 365–370 (2004).
Quinones, R. et al. Lack of achievement of a full score on the childhood myositis assessment scale by healthy four-year-olds and those recovering from juvenile dermatomyositis. Arthritis Care Res. 65, 1697–1701 (2013).
Rosina, S. et al. Development and testing of reduced versions of the manual muscle test-8 in juvenile dermatomyositis. J. Rheumatol. 48, 898–906 (2021).
Varnier, G. C. et al. Development and testing of a hybrid measure of muscle strength in juvenile dermatomyositis for use in routine care. Arthritis Care Res. 70, 1312–1319 (2018).
Campanilho-Marques, R. et al. Comparison of the utility and validity of three scoring tools to measure skin involvement in patients with juvenile dermatomyositis. Arthritis Care Res. 68, 1514–1521 (2016).
Ahmed, S., Chen, K. L. & Werth, V. P. The validity and utility of the cutaneous disease area and severity index (CDASI) as a clinical outcome instrument in dermatomyositis: a comprehensive review. Semin. Arthritis Rheum. 50, 458–462 (2020).
Craig, J., Feldman, B. M., Spiegel, L. & Dover, S. Comparing the measurement properties and preferability of patient-reported outcome measures in pediatric rheumatology: PROMIS vs CHAQ. J. Rheumatol. 48, 1065–1072 (2021).
Patel, R. N. et al. Comparison of patient-reported outcomes measurement information system computerized adaptive testing versus fixed short forms in juvenile myositis. Arthritis Care Res. 75, 381–390 (2021).
Deakin, C. T. et al. Identification and prediction of novel classes of long-term disease trajectories for patients with juvenile dermatomyositis using growth mixture models. Rheumatology 60, 1891–1901 (2021).
Aggarwal, R. et al. Autoantibody levels in myositis patients correlate with clinical response during B cell depletion with rituximab. Rheumatology 55, 991–999 (2016).
Reed, A. M. et al. Biologic predictors of clinical improvement in rituximab-treated refractory myositis. BMC Musculoskelet. Disord. 16, 257 (2015).
Tiniakou, E. et al. More severe disease and slower recovery in younger patients with anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Rheumatology 56, 787–794 (2017).
Rice, G. I. et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J. Clin. Immunol. 37, 123–132 (2017).
Lerkvaleekul, B. et al. Siglec-1 expression on monocytes is associated with the interferon signature in juvenile dermatomyositis and can predict treatment response. Rheumatology 61, 2144–2155 (2022).
Neely, J. et al. Gene expression meta-analysis reveals concordance in gene activation, pathway, and cell-type enrichment in dermatomyositis target tissues. ACR Open Rheumatol. 1, 657–666 (2019).
Bellutti Enders, F. et al. Correlation of CXCL10, tumor necrosis factor receptor type II, and galectin 9 with disease activity in juvenile dermatomyositis. Arthritis Rheumatol. 66, 2281–2289 (2014).
Wienke, J. et al. Galectin-9 and CXCL10 as biomarkers for disease activity in juvenile dermatomyositis: a longitudinal cohort study and multicohort validation. Arthritis Rheumatol. 71, 1377–1390 (2019).
Wienke, J. et al. Endothelial and inflammation biomarker profiles at diagnosis reflecting clinical heterogeneity and serving as a prognostic tool for treatment response in two independent cohorts of patients with juvenile dermatomyositis. Arthritis Rheumatol. 72, 1214–1226 (2020).
Acknowledgements
The authors would like to thank the members of the UK JDM Cohort and Biomarker Study (JDCBS) and the patients and families who contribute to it. The JDCBS is currently funded by grants from the Great Ormond Street Hospital (GOSH) Children’s Charity, National Institute for Health Research (NIHR) Biomedical Research Centre at GOSH, Cure JM, Myositis UK, Versus Arthritis and Remission Charity. M.G.L.W. is supported by grants from Cure JM, the NIHR Biomedical Research Centre at GOSH, and Versus Arthritis. The authors would also like to thank Dr S. Tansley for critical review of the manuscript. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, contributed substantially to discussion of the content, wrote the article and reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
All authors declare that the UK JDM Cohort and Biomarker Study (JDCBS) is currently funded by grants from the Great Ormond Street Hospital (GOSH) Children’s Charity, the National Institute for Health Research (NIHR) Biomedical Research Centre at GOSH, Cure JM, Myositis UK, Versus Arthritis and Remission Charity. L.R.W. declares that she is a consultant for Pfizer Inc (with consulting fees going wholly to the University College London). The other authors all declare no competing interests.
Peer review
Peer review information
Nature Reviews Rheumatology thanks L. Rider, who co-reviewed with M. Sherman; H. Kim; A. Huber; and K. Ardalan for their contribution to the peer review of this work.’
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
British Society for Rheumatology: https://rheumatology.org.uk
Childhood Arthritis and Rheumatology Research Alliance: https://carragroup.org
International Myositis Assessment & Clinical Studies Group: https://www.niehs.nih.gov/research/resources/imacs/
Juvenile dermatomyositis cohort biomarker study and repository: https://juveniledermatomyositis.org.uk/study-tools/
Paediatric Rheumatology International Trials Organisation: https://printo.it
The International Myositis Society: https://imyos.org
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Papadopoulou, C., Chew, C., Wilkinson, M.G.L. et al. Juvenile idiopathic inflammatory myositis: an update on pathophysiology and clinical care. Nat Rev Rheumatol 19, 343–362 (2023). https://doi.org/10.1038/s41584-023-00967-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41584-023-00967-9
This article is cited by
-
Anti-synthetase syndrome in a child with pneumomediastinum: a case report and literature review
BMC Pulmonary Medicine (2024)
-
Fortschritte in der Diagnostik und Therapie der juvenilen Dermatomyositis
Zeitschrift für Rheumatologie (2024)
-
Juvenile dermatomyositis: association between nail fold capillary end row loop– area under the curve– and disease damage indicators
Pediatric Rheumatology (2023)
-
Insights into juvenile myositis via engineered muscle
Nature Reviews Rheumatology (2023)
