
Abstract
Aquaporins (AQPs) are of vital importance in the cellular transport system of all living organisms. In this study, genome-wide identification, distribution, and characterization of AQPs were determined in Arabidopsis lyrata, Capsella grandiflora, C. rubella, Eutrema salsugineum, Brassica rapa, B. oleracea, and B. napus (canola). Classification and phylogeny of AQPs revealed the loss of XIPs and NIP-IIIs in all species. Characterization of distinctive AQP features showed a high level of conservation in spacing between NPA-domains, and selectivity filters. Interestingly, TIP3s were found to be highly expressed in developing seeds, suggesting their role in seed desiccation. Analysis of available RNA-seq data obtained under biotic and abiotic stresses led to the identification of AQPs involved in stress tolerance mechanisms in canola. In addition, analysis of the effect of ploidy level, and resulting gene dose effect performed with the different combinations of Brassica A and C genomes revealed that more than 70% of AQPs expression were dose-independent, thereby supporting their role in stress alleviation. This first in-depth characterization of Brassicaceae AQPs highlights transport mechanisms and related physiological processes that could be exploited in breeding programs of stress-tolerant cultivars.
Similar content being viewed by others
Introduction
Aquaporins (AQPs) are integral membrane proteins that facilitate the transport of water and many other small molecules like urea, boric acid, silicic acid, ammonia, and carbon dioxide1,2,3. AQPs are found in all living organisms and localized in the plasma membrane, endoplasmic reticulum, vacuoles, plastids and other subcellular compartments4, 5. Most AQPs are characterized by six transmembranes (TM) alpha helices connected by five loops and highly conserved NPA (Asn-Pro-Ala) motifs6. The NPA motifs are the two half TM helices forming a constrict at the center of the pore that serves as a size selectivity barrier (Törnroth-Horsefield et al., 2006). Another constrict that defines the substrate specificity consists of four amino acids known as the aromatic/arginine (ar/R) selectivity filter (SF)2, 7. In addition to these two significant selectivity barriers, several other conserved features are known to play a role in AQP solute specificity including Froger’s residues composed of five conserved amino acids known to discriminate glycerol-transporting aquaglyceroporins (GLPs) from water-conducting AQPs8. Similarly, several specificity-determining positions (SDPs) have been proposed for urea, boric acid, silicic acid, ammonia, carbon dioxide and hydrogen peroxide9.
Plant AQPs constitute a large family of proteins that are categorized, based on phylogenetic distribution, into five major sub-families: plasma membrane intrinsic proteins (PIPs), tonoplast intrinsic proteins (TIPs), NOD26-like intrinsic proteins (NIPs), small basic intrinsic proteins (SIPs), and uncharacterized intrinsic proteins (XIPs)10,11,12. Unique patterns of subcellular localization have been observed in proteins within each AQP sub-family; for instance, PIPs are mostly localized to the plasma membrane, TIPs to the tonoplast, NIPs to the plasma membrane or the endoplasmic reticulum, SIPs to the endoplasmic reticulum, and XIPs mostly to the plasma membrane4, 5. Two additional subfamilies, GlpF-like intrinsic proteins (GIPs) and hybrid intrinsic proteins (HIPs), have also been described in primitive plant species, and are thought to have been lost by higher plants during the course of evolution12, 13. Similarly, loss of XIPs in Arabidopsis and monocots, and NIP2s (categorized as NIP-III) in Arabidopsis has been confirmed in a recent study highlighting genome-wide identification and comparison of AQPs in 25 plant species12.
To date, more AQP homologs have been identified in plants compared to animals. For instance, 35 AQPs in Arabidopsis, 34 in rice, 55 in poplar and 72 in soybean have been reported compared to only 13 in the human genome12, 14. This higher and differential number of AQPs in plants is likely the result of gene duplication and higher ploidy levels in plants12, 15. The AQP duplications in the plants may have occurred either by whole genome duplication, segmental duplication, or partly by ectopic recombination, replication slippage, or retrotransposon activity. The duplicated AQPs are believed to have undergone diversification and neo-functionalization leading to the evolution of different classes of specialized functions12, 16. Recently, Shi, et al.17 analyzed gene expression dose effect using resynthesized Arabidopsis tetraploids harboring varied copies of chromosomes from model plant species Arabidopsis thaliana and Arabidopsis arenosa genomes. They observed that the genes mostly involved in cell cycle, photosynthesis, and metabolism are dosage-dependent, whereas genes involved in biotic and abiotic stress tolerance mechanisms are dosage-independent. Similar genetic resources that have a combination of genomes and diverse ploidy levels are available for Brassica species18. Brassica napus is an important oilseed crop representing the third leading source of vegetable oil in the world, after soybean and palm oil. The B. napus (an amphidiploid with chromosome n = 19) family belongs to the Cruciferae (Brassicaceae) and was developed by natural interspecific hybridization between the diploid species B. oleracea (n = 9) and B. rapa (syn. campestris, n = 10). However, very little is known about AQP evolution and gene expression dynamics in these Brassica species or other members of Brassicaceae.
Aquaporins play a significant role in plant physiological processes like cell-elongation, seed germination, osmoregulation, phloem-sap movement, stomatal and leaf-water movement, cytoplasmic homeostasis, nutrient transport, and modulation of a plant’s response to biotic and abiotic stresses19,20,21. As a result, AQPs are extensively studied to define their role in the enhancement of tolerance to abiotic stresses such as salinity, flooding, drought, heat, cold and heavy metal toxicity22,23,24,25. However, fewer efforts have been applied to understand the possible role of AQPs in alleviating biotic stresses. Recently, Tian, et al.26 evaluated the role of AQPs in the induction of disease immunity pathways in Arabidopsis. They found that Arabidopsis AQP AtPIP1;4 is responsible for transporting pathogen-induced apoplastic hydrogen peroxide (H2O2) to the cytoplasm. The role of H2O2 as a regulatory and signaling molecule has been described in many cellular processes, like photosynthesis, photorespiration, stomatal movement, cell cycle, senescence, and stress response. Permeability to H2O2 has been previously observed in several AQP subfamilies PIPs, TIPs, NIPs and XIPs27.
The ever-growing availability of plant genomes and transcriptomes offers an unprecedented access to specific gene families15. In the present study, a genome-wide identification of AQPs was performed in Arabidopsis lyrata, Capsella grandiflora, C. rubella, Eutrema salsugineum, B. rapa, B. oleracea and B. napus. The AQPs were characterized by the presence of ar/R SF, Froger’s residues, NPA motifs, and other conserved domains. Furthermore, the expression profiling of AQPs using transcriptome sequencing data available for B. napus under biotic and abiotic stress from different tissues were analyzed and compared. Results obtained from this study provide the first comprehensive classification of AQPs in Brassicacea species and highlight their importance and possible roles in stress alleviation.
Materials and Methods
Genome-wide identification of AQPs
Genomic sequences and gene annotations for A. lyrata, C. grandiflora, C. rubella, and E. salsugineum were retrieved from Phytozome (https://phytozome.jgi.doe.gov), for B. rapa, and B. oleracea from Ensembl (http://useast.ensembl.org/index.html) and for B. napus from Genoscope database (http://www.genoscope.cns.fr/brassicanapus). The NCBI command-line BLAST utilities provided in BioEdit (version 7.0.9.0) software tool was used to create local databases of the transcriptome and proteome sequences of these species28. The putative AQPs were identified with BLASTp search performed using 141 known AQPs as query sequences against the local database. The query sequences included 34 rice, 35 Arabidopsis, and 72 soybean AQPs11, 16, 29. A cut-off e-value of 10−5 was applied to identify significant matches. Top hits from the multiple matches were extracted based on the highest bit score. The BLAST hits with less than a 100 bit-score were removed. Similarly, homologs of Si-efflux transporters were identified in the seven species using BLASTp search with known efflux transporters as described by Vivancos, et al.30
Identification of NPA motifs and transmembrane domains in aquaporins
The batch mode of NCBI’s Conserved Domain Database (CDD, www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) was used for the identification of known conserved domains including NPAs. AQPs showing a single or missing NPA motif were manually examined and removed from further analysis after re-confirmation with other characteristics like protein length, TM domains, and signal peptides. Transmembrane domains were detected using TMHMM, SOSUI, and TOPCONS software tools (www.cbs.dtu.dk; http://bp.nuap.nagoya-u.ac.jp)31. The results were then manually curated for altered and missing transmembrane domains.
Construction of homology-based tertiary protein structure
Tertiary protein (3D) structures for all the newly identified and previously reported AQPs were constructed using Phyre2 protein modeling server32. ProTSAV was used for quality assessment of the 3D structures33. (http://www.scfbio-iitd.res.in/software/proteomics/protsavzip.jsp)
Structural features of AQPs
The various physical and chemical parameters for AQPs were determined using ProtParam (http://web.expasy.org/protparam/). The protein subcellular localization was predicted using CELLO sever (http://cello.life.nctu.edu.tw/). The TM domains were defined using TOPCONS, SOSUI, and TMHMM31, 34, 35. Prediction of the solute specificity was performed on the basis of dual NPA motifs, ar/R SF, Froger’s residues and homology with Arabidopsis AQPs.
Classification and phylogeny of AQPs
CLUSTALW implemented in MEGA7 was used for multiple sequence alignment of AQPs36. The multiple sequence alignment was subjected to construct the phylogenetic tree using the maximum likelihood (ML) method. The stability of branch nodes in the phylogenetic tree was measured by performing 1000 bootstraps. Based on the phylogenetic distribution, AQPs were classified into PIP, TIP, NIP, and SIP sub-families and nomenclature of the AQPs were assigned as per the similarity with Arabidopsis AQPs.
Aquaporin Co-expression network
Aquaporin expression data from the publicly available RNA-seq Bio-projects PRJNA331148, PRJNA311316, PRJNA256233 and PRJNA311067 were used to construct co-expression network. Details of the data are provided in Supplementary information 1 (Supplementary Table 1). The normalized expression data were used to determine the correlation by applying Pearson product-moment correlation coefficient (Pearson r) and correlation thresholds with lower percentile rank 5 and upper percentile rank 95 used to consider significant association. The entire list of canola AQPs was used as bit genes. The co-expression network analysis was performed with Comparative Co-Expression Network Construction and Visualization tool (CoExpNetViz)37. The network was visualized using Cytoscape V. 3.3.038.
Silicon accumulation in canola
To measure Si concentrations in canola, leaf and root samples were collected from four week-old plants growing in hydroponic solutions. Rice plants were used as positive control. For the Si + treatment, liquid potassium silicate (Kasil®, PQ Corporation) was used to obtain a maximum concentration of 1.7 mM Si, and an additional K (0–0–52) was added to the control (Si-) solution to compensate for the addition of K. Leaf and root samples were dried at 60 °C for at least two days and pulverised with a bead mill homogenizer (Omni Bead Ruptor 24, Omni International). Silicon concentrations were measured from five different plants with the X-ray fluorescence spectrometry method (Niton XL3t955 GOLDD + XRF) adapted from Vivancos et al.30.
RNA-seq data analysis
Publicly available RNA-seq data for different canola tissues and conditions were used in the present study (Supplementary Table 1). To study expression of BnAQPs under drought conditions, RNA-seq data analysis was performed using CLC Genomic Workbench. The cDNA libraries constructed for the leaf and root tissues of B. napus grown under controlled and drought conditions were sequenced using Illumina HiSeq 200039. The raw sequenced reads were processed, mapped to B. napus reference genome and the transcript abundance was normalized by the RPKM (reads per kilobase of exon per million fragments mapped) using CLC Genomic Workbench. All the parameters used for RNAseq data analysis were set to default except some changes like mapping parameters allowing for a maximum of two mismatches and the maximum of 2 hits per read. The normalized expression data for all B. napus AQPs were retrieved and analysed further in MultiExperiment Viewer (MeV_4-9-0) software tool (http://www.tm4.org/mev.html). The raw reads and analysed expression data for other studies were retrieved from the Brassica napus SRA and GEO database (http://www.ncbi.nlm.nih.gov/sra and http://www.ncbi.nlm.nih.gov/gds/). Details of Bioprojects SRA and GEO accession are provided in Supplementary Table 1. Further details of library preparation, experimental design, replication, read processing and statistical analysis are provided in Supplementary information 1.
Results
Genome-wide distribution of aquaporins in Brassicaceae
Genome-wide identification performed in seven Brassicaceae species revealed a total of 380 AQPs. The AQPs were classified following a phylogenetic distribution, and named based on the Arabidopsis nomenclature. The overall pattern of AQPs distribution within the subfamilies was uniform across all Brassicaceae species analyzed (Table 1). As previously described in Arabidopsis, PIPs represented the highest number of AQPs followed by TIPs, NIPs, and SIPs, with the exception for C. grandiflora that has nine AQPs each in TIP and NIP categories. Species within the Arabidopsis clad had roughly the same number of AQPs (Table 1). In contrast, species within the Brassica clad had approximately two to four times more AQPs, which correlates precisely with the ploidy level of their respective genomes. The number of AQPs identified in each species was correlated with the genome size and the total number of predicted genes in the genome (Fig. 1).

Relationship between number of aquaporins (AQPs), genome size and total number of predicted genes in the genomes. Total AQPs identified in Arabidopsis thaliana (At), A. lyrata (Al), Capsella grandiflora (Cg), C. rubella (Cr), Eutrema salsugineum (Es), Brassica rapa (Br), B. oleracea (Bo) and B. napus (Bn) was plotted against (A) the genome size and (B) total number of predicted genes.
Phylogenetic distribution of aquaporins in Brassicaceae
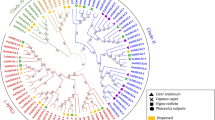
Phylogenetic analyses performed on all identified AQPs confirmed the clustering into the four distinct subfamilies PIP, TIP, NIP and SIP (Supplementary Figures S1–S7). Within each subfamily, genes from closely related species tended to cluster together (Fig. 2). The PIP subfamily divided further into two groups, PIP1 and PIP2, with PIP2 having a greater number of genes compared to PIP1 in all seven species (Supplementary Figure 8, Supplementary Table 2). The second largest subfamily, TIP, was divided into five groups. The TIP5 group was initially absent from the results obtained with genome-wide BLASTp search performed in B. oleracea genome assembly version 2.1. However, when Arabidopsis TIP5 was used as a query to perform a search in NCBI non-redundant database, the results revealed the presence of B. oleracea TIP5. The NIP subfamily contained seven groups based on the categorization previously reported in Arabidopsis. The subfamily SIP clustered into two major groups in all the analyzed genomes (Supplementary Figure 8). The subfamily XIP is entirely missing from the Brassicaceae clad.

Phylogenetic tree of aquaporins identified in seven Brassicaceae species along with the previously reported aquaporins in Arabidopsis thaliana. Aquaporins are denoted with the abbreviation of species name as At -Arabidopsis thaliana, Al - Arabidopsis lyrata, Cru - Capsella rubella, Cgr - Capsella grandiflora, Bra- Brassica rapa, Bna - Brassica napus, Bol - Brassica oleracea, Esa - Eutrema salsugineum.
Identification and localization of aquaporins on B. napus chromosomes
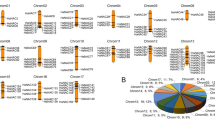
Initial results obtained with BLASTp search using a set of known AQPs identified 135 AQPs in the assembly of B. napus genome. Subsequently, based on protein length and missing transmembrane domains, 15 putative AQP-like proteins were excluded from the list (Supplementary Table 3). Localization of the 120 AQPs to B. napus chromosomes showed 49 and 45 aquaporins on genome A and C, respectively (Fig. 3). The remaining 26 AQPs were not assigned to any chromosome. However, based on the similarity with the progenitor A and C genomes (B. rapa and B. oleracea), unmapped genes were assigned to their respective genomes40. In the end, 11 and 15 AQPs were assigned to genome A random and C random, respectively, for a total of 60 AQPs on each genome. The highest number of AQPs was found on chromosomes A03 and C04, each with ten genes followed by Cnn_random with nine genes, while no AQP was found on chromosomes C9 and C10 (Fig. 3). Details about the chromosomal distribution of AQPs identified in A. lyrata, B. rapa, B. oleracea, C. grandiflora, C. rubella, and E. salsugineum are provided in Supplementary Table 4.

Chromosomal distribution of aquaporin in canola genome (Brassica napus). Random denotes the scaffolds unassigned to the chromosome. The scale is in million bases.
Gene structure of B. napus aquaporins
Large variations in intron-exon structure and organization across the four-AQP subfamilies were observed in B. napus (Fig. 4). In general, the number of introns and the length of introns/exons were found to be conserved within each group of a given AQP subfamily. Overall, the three-intron structure was commonly observed for PIPs with a few exceptions where two or four introns were present. Within the PIP2s, only BnaPIP2–5 and BnaPIP2–6 have longer introns. Another exception was found with BnaAPIP1–1c that has a longer third intron compared to closely related BnaAPIP1–1a and BnaAPIP1–1b. Interestingly, BnaASIP1–1a and BnaCSIP1–1a have 11 and 12 introns per gene respectively, a number much higher than the rest of AQPs in canola (Fig. 4).

Intron-exon structure and organization of canola aquaporin genes. Aquaporins were categorized into PIP (plasma membrane intrinsic protein), TIP (tonoplast intrinsic proteins), NIP (NOD26-like intrinsic proteins), SIP (small basic intrinsic proteins).
Physical and biochemical properties, predicted transmembrane domains, tertiary structure and subcellular localization of aquaporins
Evaluation of molecular weight (mw) and isoelectric point (pI) revealed differences among the AQP subfamilies in B. napus (Supplementary Figure 9). Aquaporin sub-families formed distinct clusters when plotted according to their mw and pI. The TIPs, besides having large variation for mw and pI, have relatively lower mw as well as pI compared to other B. napus AQP subfamilies. The TIPs have an average 26 kD mw and 5.6 pI. Similarly, B. napus PIPs are also showing a wider range with an average 29.8 kD mw and 7.6 pI. For NIPs, variation is more prominent for mw compared to pI. Most of the NIPs have pI near neutral pH. Compared to other B. napus AQP, subfamily SIP has much higher pI (average 8.8) and low mw (26.9 kD). Similar trends of variation for mw and pI were observed for AQP subfamilies in the other six Brassicaceae species (Supplementary Table 5). Most of the AQPs showed six transmembrane domains, typical hourglass tertiary structure, and plasma-membrane localization (Supplementary Dataset 1–3).
Conserved NPA motifs and ar/R selectivity filters in aquaporins
Search performed at CDD database identified two NPA signature motifs in 113 B. napus AQPs (Table 2). For the other seven AQPs, putative NPA motifs were located based on manual evaluation of protein sequence alignment. Several different amino acid compositions at NPA motifs were observed in B. napus that included: N-P-V, N-P-S, N-P-L, N-P-C and N-P-T. Amino acid sequence variation at NPA motifs was uniformly observed across AQPs identified in the seven species (Supplementary Tables 6–11). For instance, all the NIP5 homologs identified in seven species have conserved N-P-S at LB and N-P-V at LE. A NPA-NPA spacing of 108 AA was also conserved for all NIP5 proteins. However, there was significant variation in the ar/R SF positions. Another notable example is AtNIP1-1 and AtNIP1-2 both having N-P-A at LB and N-P-G at LE and 116 AA NPA-spacing. AtNIP1-1 has homologs only in A. lyrata, C. rubella, and C. grandiflora but is absent in Brassica species and E. salsugineum, whereas AtNIP1-2 has homologs in all seven species. The entire set of NIP1 homologs has 116 AA NPA-spacing except for BolCNIP1-2b where 111 AA NPA-spacing was observed.
The AAs at four ar/R SF positions are highly conserved for B. napus PIPs and TIPs, while more variations were observed with NIPs and SIPs. The entire set of 43 PIPs identified in B. napus has a F-H-T-R SF in H2, H5, LE1 and LE2. In the TIP subfamily, there is variation in ar/R SF composition across the groups. All members of B. napus TIP1s harbor H-I-A-V, TIP2s H-I-G-R, TIP4 A-I-A-R, and TIP5 N-V-G-C. TIP3s have variations at ar/R SF H5 position and contain an AA composition H-[I/M]-A-R. A distinct or common composition for ar/R SF was not observed within B. napus NIP groups. The B. napus SIP1 has two different ar/R SFs: T-V-P-I and V-F-P-I. All the SIP2s have S-H-G-A. Similar to the NPA motifs, the ar/R SFs are also conserved across all Arabidopsis AQP homologs.
A residue in Loop C (Lc) contributing to ar/R selectivity filter newly identified in Arabidopsis AtTIP2-141 was found to be conserved across all the TIPs identified in B. napus and six other species analyzed in the present study. The protein sequence alignment of TIPs from all the seven species revealed that TIP1s and TIP3s had an F residue whereas TIP2s and TIP4s had H, and TIP5s had Y at Lc ar/R SF position (Fig. 5). Similarly, the Lc ar/R SF was found to be conserved across all other subfamilies.

Alignment of Brassica napus TIPs showing 5th selectivity filter newly identified in Arabidopsis AtTIP2-1.
Absence of NIP-IIIs in Brassicaceae and Si accumulation in canola
A notable absence of specific AQPs from Brassicaceae was the group NIP2 with G-S-G-R SF pore (NIP-III), known to allow Si permeability, and often referred to as Si influx transporter or Lsi1 (Table 2). On the other hand, our genomic analysis revealed the presence of a putative Si efflux transporters (Lsi2) (Supplementary information 2). When canola plants were tested for their ability to absorb Si, results clearly showed that they did not accumulate Si above the background level (Supplementary Figure 14).
AQP expression profiling across different conditions and tissues
A total of 74 sequenced RNA-seq libraries representing five independent experiments were used to evaluate the expression profiling of B. napus AQPs (Supplementary Dataset 4 and 5). Out of 120 AQPs, 104 were found to be expressed with a criterion of >2 RPKM at least in a single library out of 74 used to claim expression of AQPs (Supplementary Dataset 4 and 5). The 16 unexpressed AQPs include 10 NIPs, four TIPs and two SIPs. On the other hand, expression was observed for all PIPs. Overall PIPs and TIPs had much higher levels of expression with an average of 154 and 187 RPKM, respectively. Comparatively, NIPs and SIPs have lower levels of expression with averages of 13 and 35, respectively. However, large variations across tissues and experimental conditions were observed for AQPs in all four subfamilies (Supplementary Dataset 4 and 5).
Expression of B. napus aquaporins in seed tissues
The RNA-seq data from Bioproject PRJNA311067 (Supplementary Table 1) were used to study expression of AQPs in seed tissues. The overall AQP activity in developing seed tissues harvested at 2, 4, 6, and 8 weeks after pollination (WAP) increased over two-fold from 415 (2 WAP) to 956 RPKM (8 WAP). The PIP and TIP subfamilies had particularly high levels of expression compared to SIPs and NIPs (Fig. 6A). Expression of PIPs, TIPs and NIPs initially increased to 4 WAP whereas SIPs expression steadily decreased over time. At four WAP, expression of PIPs went down drastically, but the expression of TIPs kept on increasing until 8 WAP (Fig. 6A). However, very high variations were observed within the subfamily, which prompted further analyses into different groups in each subfamily.

Expression of Brassica napus AQP in developing seeds. RNA-seq expression data available (Bioproject PRJNA311067) for B. napus seed at 2, 4, 6, and 8 weeks after pollination was used for AQP expression profiling. Graphs are showing expression level in term of average reads per kilobases of transcript per million mapped reads (RPKM) for (A) four AQP subfamilies namely PIP, Plasma membrane intrinsic protein; TIP, Tonoplast intrinsic protein; NIP, Noduline-26 like intrinsic protein; SIP, Small intrinsic protein; and (B) groups in each subfamily. Bars represent standard error from the mean for all values within a subfamily or a group. Detailed data are provided in Supplementary Dataset 4.
Among the different groups, PIP1s and TIP3s have the highest level of expression in seed tissue (Fig. 6B). During early development at 2 WAP, PIP1s had a fairly high level of expression whereas TIP3s had a low level. Over time as seeds matured, expression of PIP1s decreased in contrast to expression of TIP3s that increased several folds during the late stages of seed development. In the developing seeds, 86 out of the 120 AQPs were expressed with average > 2 RPKM. The remaining 34 AQPs with low or no expression in seed tissue mostly included NIPs (15) and TIPs (13). Hierarchical clustering of the AQPs expressed in the seeds showed two major clusters based on the expression pattern (Supplementary Figure 10). All TIP3 genes clustered together and showed a similar expression pattern while the other AQPs formed a separate cluster. Co-expression analysis revealed a negative correlation of TIP3s with most of the other subfamilies (Supplementary Figure 11). Similarly, a positive correlation between the PIP1s and PIP2s, and among TIP2s was observed with co-expression network analysis.
Expression of B. napus aquaporins under drought conditions
The RNA-seq data (Bioproject PRJNA256233) analysis revealed an interesting contrast for AQP expression between leaves and roots of B. napus under drought conditions. The overall expression of AQPs increased over two-fold in leaves under drought conditions while it decreased significantly in roots (Supplementary Figure 12A). This change in expression was more prominent in PIPs and TIPs (Supplementary Figure 12A). In the PIP subfamily, both PIP1s and PIP2s were particularly highly expressed. However, in the TIP subfamily, only members of the TIP1 group had a significantly higher level of expression in both tissues and under both control and drought treatments (Supplementary Figure 12B). With a cut-off >2 log2 fold change and average 2 RPKM, 16 AQPs were identified as differentially expressed genes (DEGs) in leaves. All of these genes were up-regulated under the drought treatment. Interestingly, all 16 DEGs belong to PIP and TIP subfamilies.
Expression of B. napus aquaporins under biotic stress
Expression of B. napus AQPs under biotic stress was evaluated using the RNA-seq transcriptome data (Bioproject PRJNA311316) available for the hemibiotrophic fungal pathogen Leptosphaeria maculans inoculated to resistant (LepR1) genotype DF78 and the susceptible genotype Westar. In the resistant genotype, the number of differential expressed AQPs was much higher compared to the susceptible genotype. More specifically, PIPs were particularly highly expressed in the resistant genotype (Fig. 7A). Most of the PIP1s and PIP2s were up-regulated during the early stage, but expression was reduced drastically during the later stage of disease development in resistant genotype Df78. In the case of TIPs, only TIP1s has significantly higher expression. Interestingly, in susceptible genotype Westar, TIP1 expression was higher at 3 dpi but receded during later stages (Fig. 7B). In contrast, the highest level of TIP1 expression was observed at 7 dpi in Df78 genotype. Among the NIPs and SIPs, NIP6s and SIP2s had higher expression levels.

Expression of Brassica napus AQP in susceptible and resistant genotypes after inoculation with Leptosphaeria maculans retrieved from Bioproject PRJNA311316. Graphs are showing expression level in term of average reads per kilobases of transcript per million mapped reads (RPKM) for (A) four AQP subfamilies namely PIP, Plasma membrane intrinsic protein; TIP, Tonoplast intrinsic protein; NIP, Noduline-26 like intrinsic protein; SIP, Small intrinsic protein; and (B) groups in the each subfamily. Bars represent standard error from the mean for all values within a subfamily or a group out of three biological replicates. Detailed data are provided in Supplementary Dataset 4.
Another set of RNA-seq data (Bioproject PRJNA331148) publicly available for B. napus infected with the necrotrophic fungus Sclerotinia sclerotiorum (Ssp) and the biocontrol agent Pseudomonas chlororaphis (PA23) was used to further investigate AQP expression dynamics. Comparison of AQP expression in mature leaf tissue inoculated with water, Ssp ascospores, PA23, or both PA23 and Ssp (PASs), revealed much lower expression levels of AQPs following Ssp infection (Fig. 8A). By contrast, PIPs and TIPs had higher levels of expression in control and PA23-inoculated samples. In the case of samples inoculated with both PA23 and Ssp, expression of PIPs and TIPs was slightly lower compared to controls but still much higher than in the samples infected with Ssp alone (Fig. 8A). Overall, PIP1s, PIP2s, TIP1s and TIP2s were the most expressed AQPs (Fig. 8B). Interestingly, SIPs contrasted with the other groups both in levels and patterns of expression according to treatments (Fig. 8B). A total of three clusters were formed with hierarchical clustering (Supplementary Figure 13). Cluster 1 grouped AQPs with the highest expression in control, followed by PA23, and PASs, and lowest in Ssp; Cluster 2 represented AQPs with high expression in control and PA23, slightly lower in PASs, and much lower in Ssp; finally, Cluster 3 grouped AQPs with low overall expression.

Expression of Brassica napus AQP in mature leaf inoculated with Sclerotinia sclerotiorum (Ssp), biocontrol agent Pseudomonas chlororaphis (PA23) and combined both together retrieved from Bioproject PRJNA331148. Graphs are showing expression level in term of average reads per kilobases of transcript per million mapped reads (RPKM) for (A) four AQP subfamilies namely PIP, Plasma membrane intrinsic protein; TIP, Tonoplast intrinsic protein; NIP, Noduline-26 like intrinsic protein; SIP, Small intrinsic protein; and (B) groups in each subfamily. Bars represent standard error from the mean for all values within a subfamily or a group out of three biological replicates. Detailed data are provided in Supplementary Dataset 4.
Aquaporin expression with different levels of gene dosage
The transcriptome profiling data (Bioproject PRJNA322687) available for B. rapa (A genome, 2n = 20) and B. oleracea (B genome, 2n = 18) parents and their synthesized progenies with different copies of sub-genomes (AC, AAC, CCA, CCAA) were used to evaluate gene dosages effect on AQP expression18. Overall triploid hybrid CCA (2n = 28) and AAC (2n = 29) showed significantly higher AQP expression compared to diploid parental lines, B. napus (AC, 2n = 19) and allotetraploid CCAA (2n = 38) (Fig. 9A). The AA genotype has higher expression than the CC genotype and this was also consistent with the triploids where AAC had greater AQP expression than CCA. Interestingly, the AQP expression in CCAA was much higher than in AC despite having equivalent fractions of A and C genomes. The expression of PIP and TIP subfamily was more affected by ploidy levels and by genome combinations (Fig. 9A).

Expression of aquaporins (AQPs) in B. rapa (A genome, 2n = 20) and B. oleracea (B genome, 2n = 18) parents and their synthesized progenies with different copies of sub-genomes (AC, AAC, CCA, CCAA) retrieved from Bioproject PRJNA322687. Graphs are showing expression level in term of average reads per kilobases of transcript per million mapped reads (RPKM) for (A) four AQP subfamilies namely PIP, Plasma membrane intrinsic protein; TIP, Tonoplast intrinsic protein; NIP, Noduline-26 like intrinsic protein; SIP, Small intrinsic protein; and (B) groups in each subfamily. RNA-seq expression data was obtained from Tan et al. (2016). Bars represent standard error from the mean for all values within a subfamily or a group out of six samples from two biological replicates. Detailed data are provided in Supplementary Dataset 5.
Comparison within the groups across all the subfamilies showed overall similar trends, with the highest expression in AA followed by CC and then AC (Fig. 9B). Similarly, the highest level of expression was observed in AAC followed by CCA and CCAA. An exception to this common trend was found in TIP2, SIP1 and SIP2. In the case of SIPs, the descending order for expression level was AC, AA and then CC. In triploids and tetraploids, the descending order for expression of SIP1 was CCAA, AAC and CCA but SIP2 had higher expression in CCAA followed by CCA (Fig. 9B).
Dosage-dependent genes were identified by Pearson’s correlation analysis performed between the expression data and genome composition. For instance, AA, AC, AAC, CCA, and CCAA have A genome composition as 1, 0.5, 0.66, 0.33, and 0.5 respectively. About 98% of the AQPs showed a positive correlation with the genome composition (Supplementary Dataset 5). Only 30% of expressed AQPs were significantly correlated with genome composition (p < 0.01, Supplementary Dataset 5). The AQPs assigned to A genome were more dosage dependent (p < 0.01) compared to AQPs on C genome. PIPs, including both PIP1s and PIP2s, showed more dosage dependent patterns of expression. In the TIP subfamily, only TIP1s, and in SIPs only SIP1s showed gene dosage dependency. For their part, NIPs showed mostly dosage-independent expression.
Discussion
In the present study, genome-wide identification and characterization of AQPs were performed in Brassicaceae species to understand their evolution within this important group of plants and to further analyze their possible molecular role, particularly in B. napus. The first genome-wide AQP study was carried out in Arabidopsis and served as a reference for the identification of homologs in many other crop plants15, 20, 21. Being a member of the Brassicaceae family, Arabidopsis provides a close phylogenetic resource to infer information for economically important crop species in the family. To perform the comparative analysis, we have exploited genomic information available for species from two closely related tribes: the Camelineae including A. thaliana, A. lyrata, C. rubella, C. grandiflora, and the Calepineae including B. rapa, B. napus, B. oleracea and E. salsugineum. These genomes together with available transcriptomic resources have made it possible to understand many aspects including genomic distribution, structural and functional conservation, phylogenetic relations and the expression dynamics across different tissues and environmental conditions.
Loss of XIPs and silicon transporter NIPs in Brassicaceae
Genomic distribution of AQPs have been studied in several plant species including monocots, dicots and primitive species like Physcomitrella patens, Picea abies, Selaginella moellendorffii12. The primitive species have two more AQP subfamilies: GlpF-like intrinsic protein (GIPs) and hybrid intrinsic protein (HIPs), along with the five commonly observed families in higher plants. As in other higher plants, the two subfamilies HIP and GIPs are absent in Brassicaceae. The XIP family, common to most of the dicots, are also missing in all studied species inferring a recent loss during evolution (Supplementary Table 12). The XIPs have been reported to transport several solutes like glycerol, H2O2, copper, boric acid, and arsenic in tobacco42. Similarly, Noronha, et al.43 have reported a role for XIPs in drought stress and also in the transport of glycerol, hydrogen peroxide, heavy metals in grapevine. The loss of XIPs in Brassicaceae suggest that the functions of XIPs were shared with other AQPs or were of lesser importance.
Another interesting feature in Brassicaceae was the absence of NIPs with G-S-G-R SF (NIP-III) known to be essential for silicon (Si) transport. Recent evidence has linked the presence of those NIPs with, Si accumulation and benefits from Si fertilization in many plant families. Brassicaceae now join Solanaceae among families that are poor Si accumulators on the basis of AQP profile (Supplementary Figure 14)12. This is further supported by our phenotyping results showing clearly that canola could not accumulate Si, and previous reports44, 45 that showed that Arabidopsis was a poor Si accumulator. Interestingly, putative Si-efflux transporters were identified in all seven species, which would suggest that the presence or absence of Si permeable NIP-IIIs is the critical factor in determining the ability of a plant to absorb Si.
Recent genome duplications attributed to high number of AQPs in Brassicaceae species
In spite of important variations in genome size, a comparable number of AQPs were identified among species in the Camelineae tribe. For instance, the A. lyrata genome is almost double the size of Arabidopsis and yet has an overall similar number of AQPs46. Genome size variation in these closely related species is attributed to large-scale rearrangements, including many small deletions in the non-coding region and transposons46. On the other hand, in the four Calepineae species, variations observed in the total number of AQPs are attributed more to variations in genome size resulting from polyploidization. Overall the number of AQPs were well correlated with genome size and the total number of genes, an observation consequent with the relatively recent duplication events in these species40. Interestingly, the observed correlation contradicts earlier reports where a negative correlation was observed between the percentage of protein-coding genes and the genome size47. For instance, the B. napus genome, known to be formed by recent allopolyploidy between B. rapa (AA) and B. oleracea (CC), has a number of AQPs equal to the total number of AQPs found in both species. The number of AQPs in each subfamily corresponding to AA and CC genome of B. napus also correlates well with the respective ancestral genomes (B. oleracea and B. rapa). Most of the B. napus AQPs identified here were mapped on assembled chromosomes except for 26 AQPs found in random scaffold regions of perturbed synteny (see Fig. 3). This could lead to a possible change in total number of AQPs with improved genome assembly in the future.
Gene structure, biochemical and physical properties, and conserved domains in aquaporins
Intron-exon organization is necessary to confirm the polyphyletic origin of true gene homologs and paralogs and to understand the functionality of the gene43, 48. The intron-exon organization was found to be conserved within each AQP subfamily and more prominently among the genes paired phylogenetically. Such conserved intron-exon organization of AQP subfamilies has also been observed in several plant genomes including chickpea, soybean, rice, and Arabidopsis 16, 21. Similarly, biochemical and physical properties were also found to be well aligned with the phylogenetic distribution. Molecular weight and pI are also indicative of the subcellular localization. Earlier, proteomic analysis revealed relatively lower pI (6.69) for vacuolar proteins as compared to all other proteins (pI 7.40) in the Arabidopsis genome49. This is consistent with the lower pI predicted in the present study for the TIPs (pI 5.6).
The NPA signature motif observed in AQPs are known to have a role in the regulation of solute transport and also in the localization of the protein in the membrane50, 51. In the present study, only the third AA position of the NPA motif showed variation in some of the NIPs and SIPs. This suggests a lesser selectivity role for the third amino acid in these subfamilies. A mutagenesis study performed with AtNIP5;1 showed no significant effect on the transport of solutes (Si and arsenate) by changing the original NPS and NPV with the NPA motif 52. However, mutagenesis experiments performed with Si transporter AQPs revealed that the conserved NPA-NPA spacing played a crucial factor in substrate selectivity12. This might be similarly critical for other AQP subfamilies since the spacing between NPA motifs were found to be conserved for homologs across the seven species. In the case of Ar/R SF, apart from having considerable variation among the subfamilies and groups, it was found to have a high level of conservation among homologs. This suggests a possible diversity of solute transport by the AQPs and the involvement of the conserved Ar/R SFs in the process. In plants, only a few protein crystallographic studies are available and the current understanding of four Ar/R SF is mostly based on AQPs from animal and microbial origin15. Interestingly, a newly reported fifth AA, involved in the formation of SF in AtTIP2-141, was observed to be conserved in TIPs and all other AQPs suggesting the possibility of five AA SF involved in solute permeability in plant AQPs.
Tissue specificity of the AQPs
Very limited efforts have been deployed to understand tissue specificity of genome-wide AQPs in plants. Ubiquitous expression in roots and leaves for PIPs and TIPs, the two subfamilies with consistently higher expression, have been widely reported. However, other crucial stages for crop yield such as seed development have not been well studied. In this context, transcriptomic analysis during seed development provides new insights into AQPs role. In early developmental stages, upregulation of many PIPs and TIPs in seeds suggests an active growth and a higher level of water and nutrient movement in the tissues, whereas downregulation of most PIPs and TIPs (except for one TIP3) was observed as the seed matured, an observation consistent with the fact that water content of the seed reduces drastically during the maturation stage. In the same manner, the only TIP3 which was found continuously upregulated is thought to have a role in seed desiccation and in lipid metabolic pathways16, 53. Earlier, a similar type of expression pattern for TIPs and more particularly TIP3s was reported in soybean16. Given that both B. napus and soybean have high seed oil content, this suggests a key role of TIP3s in regulating oil content of oilseed crops. Negative correlation of TIP3s with other subfamilies is expected because of their tissue specificity and higher expression in seeds.
Expression of AQPs under abiotic stress
Differential expression of AQPs has been studied under several abiotic stress conditions including salinity, drought, and extreme temperature in many plant species54, 55. Most of the previous studies were performed with a limited number of AQPs using quantitative PCR expression profiling. As such, expression profiling covering all the canola AQPs provides a better genome-wide understanding of expression dynamics under water limiting environments. In previous efforts, a very conflicting pattern of AQP expression was observed in different plant species (as reviewed in Forrest and Bhave, 2007). Under drought stress, both up and down regulation of AQPs in root and leaves have been reported with no clear delineation of specific roles linked to precise AQPs. In the present study, contrasting AQP expression with notable downregulation in roots and upregulation in leaves was highlighted. It is well established that the pattern of AQPs expression can vary drastically with the magnitude of the stress56. For example, in Vitis hybrids, a severe stress decreased the overall expression of AQPs initially, but, following continuous stress, the expression of AQPs significantly increased in leaves and remained low in roots56. In another report, transgenic Arabidopsis plants overexpressing PIP1;4 or PIP2;5 showed a rapid water loss and became susceptible to water stress57. A similar case of overexpression of PIP1s has been observed in tobacco58. The reduced expression of AQPs is believed to prevent loss of metabolic energy under severe stress situations and/or prevent water loss from the root to the hypertonic surrounding environment. The contrasting response of AQPs in leaves and roots may also be attributed to different roles of AQPs in different tissues59.
Aquaporins expression as an indicator of biotic stress
Plant pathogen development depends on the host for nutrition and water. Therefore, water movement at sites of infection is crucial for disease progress (Nature last week). However, AQPs have rarely been considered as candidate players in biotic stress studies. Most of the transcriptomic studies employing dual RNA-seq focus on pathogenicity-related genes and their host counterpart resistance genes and disease response genes60,61,62. In a recent pioneer study by Tian, et al.26, the group showed that an Arabidopsis aquaporin (AtPIP1;4) was involved in the transport of H2O2 and subsequent induction of disease immunity pathways. This report highlights convincingly the potential role of AQPs during biotic stress conditions. In the present study, analysis of dual RNA-seq data available for L. maculans inoculated in resistant canola genotype DF78 and susceptible genotype Westar showed high expression of PIPs exclusively in the resistant genotype (see Fig. 7). In a previous study, Mysore, et al.63 showed that overexpression of disease resistance gene Pto in line with upregulation of AQP genes, was more prominent after pathogen infection. A similar type of response was observed here with resistant genotype DF78 where expression of AQPs was upregulated in the early stage of infection. Overall it would appear that under disease stress, the expression of AQPs is affected as shown by the lower expression of PIP2s in a susceptible cultivar. However, it is hard to pinpoint the causal gene(s) and associated mechanisms involved in disease resistance. However, the response of AQPs under biotic stress is easier to separate with the analyses performed here with another set of transcriptomic data involving a tripartite interaction host-pathogen and biocontrol agent. As with infection with L. maculans, the AQP expression was downregulated compared to controls in B. napus plants infected with S. sclerotiorum (Ssp), but was less affected when the plants were inoculated with the biocontrol agent P. chlororaphis (PA23). Accordingly, a reduced AQP expression seems to be an indicator of biotic stress in plants. Further studies are needed to elucidate the direct role of AQPs in biotic stress regulation.
Dosage-dependency of aquaporin homologs
Understanding of dosage-dependency of gene expression is important to predict several attributes of molecular regulations and functionality. Tan, et al.18 suggested that dosage-dependent genes were involved in the basic biological processes, such as growth and development, whereas dosage independent ones were likely involved in stress responses. In the present study, by analyzing available RNA-seq data, only 30% of AQPs showed dosage-dependency, suggesting a large proportion of genes involved in stress tolerance mechanisms. Furthermore, Tan, et al.18 reported that dosage-independent genes were more likely to take part in protein–protein interactions. This would mean that dosage-independent NIPs and most of the TIPs are probably involved in protein-protein interactions. In addition, information provided about dosage-dependency is also helpful to predict involvement of trans or cis regulation of gene expression18.
Conclusions
Genome-wide identification and analysis of AQPs performed here in seven Brassicaceae species highlighted several novel findings explaining their evolution and functional regulation. By taking advantage of the first fully sequenced polyploid genome of B. napus, we have shown that recent genome-wide duplications resulted into a higher number of AQPs with practically no loss after duplication. This translated into showing that B. napus has the highest number of AQPs in a plant species to date. Expression analysis showed similar patterns of tissue-specific expression for B. napus AQPs as previously observed in soybean, rice and Arabidopsis. Analysis of AQPs expression in developing seeds suggested the key role of TIP3s in seed maturation. The study of TIP3s is particularly important in oilseed crops since maturation and desiccation greatly influences seed oil content. Furthermore, extensive analysis of AQPs under abiotic and biotic stress highlighted the involvement of AQPs in stress tolerance mechanisms. More particularly, analysis of infected plants treated with a biocontrol agent showed a lowered expression of AQPs, a phenomenon that opens leads to understand plant physiology under disease conditions. RNA-seq transcriptome data also helped to identify dosage-dependent and independent AQPs in canola that has relevance in understanding protein-protein interactions, cis or trans regulation, stress mechanisms and also in the planning of transgenic experiments where effects of overexpression can be predicted. The identification, classification, evolution and functional regulation of AQPs performed in the present study will be helpful for enhancing our understanding of AQPs and for the development of more sustainable stress tolerant crops.
References
Kitchen, P. et al. Beyond water homeostasis: diverse functional roles of mammalian aquaporins. Biochimica et Biophysica Acta (BBA)-General Subjects 1850, 2410–2421 (2015).
Kreida, S. & Törnroth-Horsefield, S. Structural insights into aquaporin selectivity and regulation. Curr. Opin. Struct. Biol. 33, 126–134 (2015).
Deshmukh, R. & Bélanger, R. R. Molecular evolution of aquaporins and silicon influx in plants. Funct. Ecol. 30, 1277–1285, doi:10.1111/1365-2435.12570 (2016).
Maurel, C., Santoni, V., Luu, D.-T., Wudick, M. M. & Verdoucq, L. The cellular dynamics of plant aquaporin expression and functions. Curr. Opin. Plant Biol. 12, 690–698 (2009).
Wudick, M. M., Luu, D. T. & Maurel, C. A look inside: localization patterns and functions of intracellular plant aquaporins. New Phytol. 184, 289–302 (2009).
Jung, J. S., Preston, G. M., Smith, B. L., Guggino, W. B. & Agre, P. Molecular structure of the water channel through aquaporin CHIP. The hourglass model. J. Biol. Chem. 269, 14648–14654 (1994).
Törnroth-Horsefield, S. et al. Structural mechanism of plant aquaporin gating. Nature 439, 688–694 (2006).
Froger, A., Thomas, D., Delamarche, C. & Tallur, B. Prediction of functional residues in water channels and related proteins. Protein Sci. 7, 1458–1468 (1998).
Hove, R. M. & Bhave, M. Plant aquaporins with non-aqua functions: deciphering the signature sequences. Plant Mol. Biol. 75, 413–430 (2011).
Jang, J. Y., Kim, D. G., Kim, Y. O., Kim, J. S. & Kang, H. An expression analysis of a gene family encoding plasma membrane aquaporins in response to abiotic stresses in Arabidopsis thaliana. Plant Mol. Biol. 54, 713–725 (2004).
Quigley, F., Rosenberg, J. M., Shachar-Hill, Y. & Bohnert, H. J. From genome to function: the Arabidopsis aquaporins. Genome Biol. 3, 1 (2001).
Deshmukh, R. K. et al. A precise spacing between the NPA domains of aquaporins is essential for silicon permeability in plants. The Plant Journal 83, 489–500 (2015).
Danielson, J. Å. & Johanson, U. Unexpected complexity of the aquaporin gene family in the moss Physcomitrella patens. BMC Plant Biol 8, 1 (2008).
Ishibashi, K., Hara, S. & Kondo, S. Aquaporin water channels in mammals. Clin. Exp. Nephrol. 13, 107–117 (2009).
Deshmukh, R., Sonah, H. & Belanger, R. Plant Aquaporins: genome-wide identification, transcriptomics, proteomics, and advanced analytical tools. Frontiers in Plant Science 7, doi:10.3389/fpls.2016.01896 (2016).
Deshmukh, R. K. et al. Identification and functional characterization of silicon transporters in soybean using comparative genomics of major intrinsic proteins in Arabidopsis and rice. Plant Mol. Biol. 83, 303–315 (2013).
Shi, X., Zhang, C., Ko, D. K. & Chen, Z. J. Genome-wide dosage-dependent and-independent regulation contributes to gene expression and evolutionary novelty in plant polyploids. Mol. Biol. Evol. 32, 2351–2366 (2015).
Tan, C. et al. Genome-wide gene/genome dosage imbalance regulates gene expressions in synthetic Brassica napus and derivatives (AC, AAC, CCA, CCAA). Frontiers in Plant Science 7, 1432 (2016).
Li, G., Santoni, V. & Maurel, C. Plant aquaporins: roles in plant physiology. Biochimica et Biophysica Acta (BBA)-General Subjects 1840, 1574–1582 (2014).
Song, L. et al. Soybean TIP Gene Family Analysis and Characterization of GmTIP1;5 and GmTIP2;5 Water Transport Activity. Frontiers in Plant Science 7, doi:10.3389/fpls.2016.01564 (2016).
Deokar, A. A. & Tar’an, B. Genome-Wide Analysis of the Aquaporin Gene Family in Chickpea (Cicer arietinum L.). Frontiers in Plant Science 7, doi:10.3389/fpls.2016.01802 (2016).
Gao, Y.-P., Young, L., Bonham-Smith, P. & Gusta, L. V. Characterization and expression of plasma and tonoplast membrane aquaporins in primed seed of Brassica napus during germination under stress conditions. Plant Mol. Biol. 40, 635–644 (1999).
Yu, Q., Hu, Y., Li, J., Wu, Q. & Lin, Z. Sense and antisense expression of plasma membrane aquaporin BnPIP1 from Brassica napus in tobacco and its effects on plant drought resistance. Plant Sci. 169, 647–656 (2005).
Zhang, Y., Wang, Z., Chai, T., Wen, Z. & Zhang, H. Indian mustard aquaporin improves drought and heavy-metal resistance in tobacco. Mol. Biotechnol. 40, 280–292 (2008).
Sun, H., Li, L., Lou, Y., Zhao, H. & Gao, Z. Genome-wide identification and characterization of aquaporin gene family in moso bamboo (Phyllostachys edulis). Mol. Biol. Rep. 43, 437–450 (2016).
Tian, S. et al. Plant Aquaporin AtPIP1; 4 Links Apoplastic H2O2 Induction to Disease Immunity Pathways. Plant Physiol., pp. 01237.02015 (2016).
Bienert, G. P. & Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochimica et Biophysica Acta (BBA)-General Subjects 1840, 1596–1604 (2014).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symp. Ser. 95–98 (1999).
Sakurai, J., Ishikawa, F., Yamaguchi, T., Uemura, M. & Maeshima, M. Identification of 33 rice aquaporin genes and analysis of their expression and function. Plant Cell Physiol. 46, 1568–1577 (2005).
Vivancos, J. et al. Identification and characterization of silicon efflux transporters in horsetail (Equisetum arvense). J. Plant Physiol. 200, 82–89 (2016).
Bernsel, A., Viklund, H., Hennerdal, A. & Elofsson, A. Topcons: consensus prediction of membrane protein topology. Nucleic Acids Res., gkp363 (2009).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Singh, A., Kaushik, R., Mishra, A., Shanker, A. & Jayaram, B. ProTSAV: A protein tertiary structure analysis and validation server. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1864, 11–19 (2016).
Hirokawa, T., Boon-Chieng, S. & Mitaku, S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics 14, 378–379 (1998).
Krogh, A., Larsson, B., Von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol., msw054 (2016).
Tzfadia, O. et al. CoExpNetViz: comparative co-expression networks construction and visualization tool. Frontiers in Plant Science 6, 1194 (2016).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Liu, C. et al. Comparative analysis of the Brassica napus root and leaf transcript profiling in response to drought stress. Int. J. Mol. Sci. 16, 18752–18777 (2015).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014).
Kirscht, A. et al. Crystal Structure of an Ammonia-Permeable Aquaporin. PLoS Biol. 14, e1002411 (2016).
Bienert, G. P., Bienert, M. D., Jahn, T. P., Boutry, M. & Chaumont, F. Solanaceae XIPs are plasma membrane aquaporins that facilitate the transport of many uncharged substrates. The Plant Journal 66, 306–317 (2011).
Noronha, H. et al. The grapevine uncharacterized intrinsic protein 1 (VvXIP1) is regulated by drought stress and transports glycerol, hydrogen peroxide, heavy metals but not water. PLoS One 11, e0160976 (2016).
Montpetit, J. et al. Cloning, functional characterization and heterologous expression of TaLsi1, a wheat silicon transporter gene. Plant Mol. Biol. 79, 35–46 (2012).
Vivancos, J., Labbé, C., Menzies, J. G. & Bélanger, R. R. Silicon‐mediated resistance of Arabidopsis against powdery mildew involves mechanisms other than the salicylic acid (SA)‐dependent defence pathway. Mol. Plant Pathol. 16, 572–582 (2015).
Hu, T. T. et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–481 (2011).
Gregory, T. R. Synergy between sequence and size in large-scale genomics. Nature Reviews Genetics 6, 699–708 (2005).
Deshmukh, R. K., Sonah, H. & Singh, N. K. Intron gain, a dominant evolutionary process supporting high levels of gene expression in rice. J. Plant Biochem. Biotechnol. 1–5 (2015).
Carter, C. et al. The vegetative vacuole proteome of Arabidopsis thaliana reveals predicted and unexpected proteins. The Plant Cell 16, 3285–3303 (2004).
Eriksson, U. K. et al. Subangstrom resolution X-ray structure details aquaporin-water interactions. Science 340, 1346–1349 (2013).
Guan, X. G. et al. NPA motifs play a key role in plasma membrane targeting of aquaporin‐4. IUBMB life 62, 222–226 (2010).
Mitani-Ueno, N., Yamaji, N., Zhao, F.-J. & Ma, J. F. The aromatic/arginine selectivity filter of NIP aquaporins plays a critical role in substrate selectivity for silicon, boron, and arsenic. J. Exp. Bot. 62, 4391–4398 (2011).
Mao, Z. & Sun, W. Arabidopsis seed-specific vacuolar aquaporins are involved in maintaining seed longevity under the control of Abscisic Acid Insensitive 3. J. Exp. Bot., erv244 (2015).
Forrest, K. L. & Bhave, M. Major intrinsic proteins (MIPs) in plants: a complex gene family with major impacts on plant phenotype. Functional & Integrative Genomics 7, 263–289 (2007).
Afzal, Z., Howton, T., Sun, Y. & Mukhtar, M. S. The Roles of Aquaporins in Plant Stress Responses. Journal of Developmental Biology 4, 9 (2016).
Galmés, J. et al. Aquaporin expression in response to different water stress intensities and recovery in Richter-110 (Vitis sp.): relationship with ecophysiological status. Planta 226, 671–681 (2007).
Jang, J. Y. et al. Transgenic Arabidopsis and tobacco plants overexpressing an aquaporin respond differently to various abiotic stresses. Plant Mol. Biol. 64, 621–632 (2007).
Aharon, R. et al. Overexpression of a plasma membrane aquaporin in transgenic tobacco improves plant vigor under favorable growth conditions but not under drought or salt stress. The Plant Cell 15, 439–447 (2003).
Reinhardt, H. et al. Tonoplast aquaporins facilitate lateral root emergence. Plant Physiol. pp. 01635.02015 (2016).
Sonah, H. et al. Comparative Transcriptomic Analysis of Virulence Factors in Leptosphaeria maculans during Compatible and Incompatible Interactions with Canola. Frontiers in Plant Science 7, doi:10.3389/fpls.2016.01784 (2016).
Sonah, H., Deshmukh, R. K. & Bélanger, R. R. Computational Prediction of Effector Proteins in Fungi: Opportunities and Challenges. Frontiers in Plant Science 7, doi:10.3389/fpls.2016.00126 (2016).
Westermann, A. J., Gorski, S. A. & Vogel, J. Dual RNA-seq of pathogen and host. Nature Reviews Microbiology 10, 618–630 (2012).
Mysore, K. S., D’Ascenzo, M. D., He, X. & Martin, G. B. Overexpression of the disease resistance gene Pto in tomato induces gene expression changes similar to immune responses in human and fruitfly. Plant Physiol. 132, 1901–1912 (2003).
Acknowledgements
The authors would like to thank the Natural Sciences and Engineering Research Council of Canada (NSERC), the Agri-Innovation Program Growing Forward 2, SaskCanola, Agriculture and Agri-Food Canada and the Canada Research Chairs Program to RRB for financial support.
Author information
Authors and Affiliations
Contributions
H.S., R.D., C.L. and R.B. compiled and analyzed the data, drew the conclusions and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sonah, H., Deshmukh, R.K., Labbé, C. et al. Analysis of aquaporins in Brassicaceae species reveals high-level of conservation and dynamic role against biotic and abiotic stress in canola. Sci Rep 7, 2771 (2017). https://doi.org/10.1038/s41598-017-02877-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-02877-9
This article is cited by
-
Plant biomarkers as early detection tools in stress management in food crops: a review
Planta (2024)
-
Influence of Silicic Acid Foliar Spray on Foraging Behaviour of Bee Pollinators and Yield of Rapeseed
Silicon (2024)
-
Aquaporin water channels: roles beyond renal water handling
Nature Reviews Nephrology (2023)
-
Cloning and functional analysis of GhDFR1, a key gene of flavonoid synthesis pathway in naturally colored cotton
Molecular Biology Reports (2023)
-
Genome-wide identification of Fagus sylvatica aquaporins and their comparative spring and summer expression profiles
Trees (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
